Note
A simple method for modeling amyloid kinetics featuring position biased fiber breakage
2020 Volume 17 Pages 30-35

Details
2020 Volume 17 Pages 30-35
A mathematical model of amyloid fiber formation is described that is able to simply specify different rates of fiber breakage at internal versus end regions. This Note presents the derivation of the relevant equations and provides results showing the dramatic effects of position biased fiber breakage on the kinetics of amyloid growth.
A kinetic model of amyloid formation is presented based on two inter-related ordinary differential equations. The model can simply account for different rates of breakage at the fiber ends vs. internal regions. In the former case, breakage releases a monomer unable to maintain its amyloid structure. In the latter case, breakage produces two smaller fibers each able to act as ‘seeds’ able to facilitate amyloid growth. Thus position dependent susceptibility to breakage could determine whether breakage encourages or discourages amyloid growth. These outcomes are crucial when considering the role amyloid growth plays in disease aetiology, such as for Alzheimer’s disease.
Protein helical polymerization reactions, such as those constituted by actin and tubulin assembly, have been a much studied topic in biophysics [1,2]. Amyloid, a fibril-like homopolymer capable of being formed from many different protein monomer building blocks, has been shown to exhibit both helical polymerization kinetics and steady state behavior capable of quantitative description using helical polymerization models of Oosawa and colleagues [3–6]. A number of important extensions to these models been developed over the last twenty years to better describe amyloid formation [7–14]. These extensions are based on a consensus chemical schema (Fig. 1) featuring the following three general processes, (i.) Reversible fiber nucleation; (ii.) Fiber growth and dissolution via monomer addition and loss; and (iii.) Fiber breakage and joining. A simple kinetic description containing some aspects of the consensus mechanism was developed by Smith, J. F., et al. [15] who employed an approach cast in terms of average quantities. Although providing less information about the time dependent evolution of the polymer distribution, this reduced model proved particularly simple to code and produced easily interpretable output. However, due to the approximations involved in its derivation, the Smith, J. F., et al. [15] model lacked the flexibility to treat biologically relevant limiting cases of amyloid fiber growth. One such important case, described by Hall and Edskes [9], was that relating to differential rates of amyloid fiber breakage from the fiber ends (to release a monomer), versus that occurring internally (to produce two shorter amyloid fibers). A recent approach based on the method of moments [16], developed a relatively complex solution to the simulation of this differential end versus internal breakage problem by approximating the evolving amyloid polymer distribution through the use of a large number (>100) of Gaussian functions. In this Letter, I derive a simpler approximate solution to the case of position dependent fiber breakage based on the assumption of a single distribution characterizing the fibril population. The current approach retains the relative simplicity of the earlier Smith, J. F., et al. [15] model without having to resort to the complex function approach adopted by Nicoud, L., et al. [16]. The method has the added benefit that it can also simply treat cases relating to differential rates of fiber end-to-end joining.
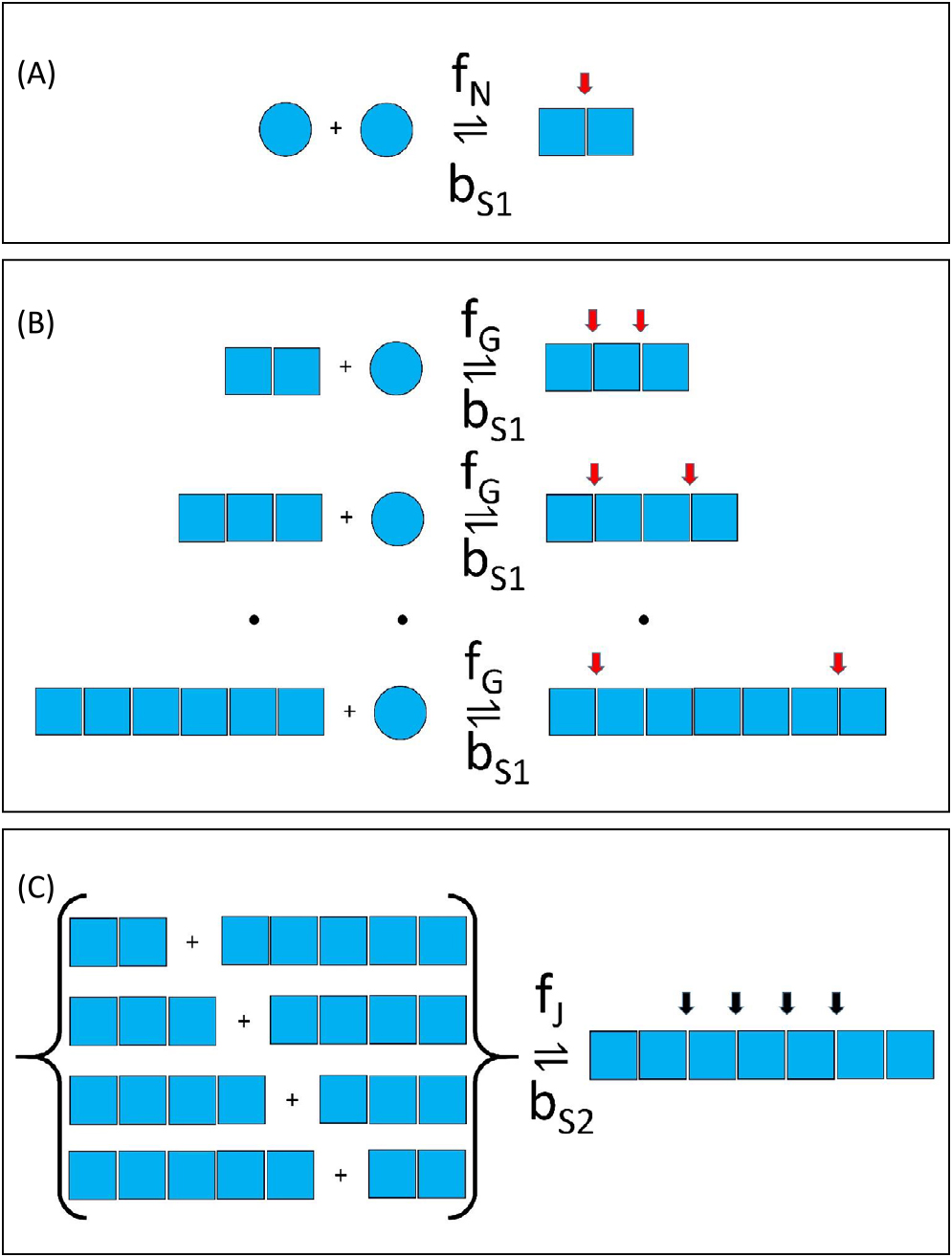
Consensus amyloid formation mechanism featuring position dependent bias in fiber breakage: (A) Nucleation—modeled as the interaction of two monomers to form a dimer nucleus with the process regulated by a second-order nucleation rate constant, fN, (units of M–1s–1) and a reverse unimolecular process governed by a first-order breakage rate constant, bS1 (units of s–1). (B) Growth and dissolution—in which fibrils can increase in size via monomer addition, regulated by a second-order growth rate constant, fG (units of M–1s–1) and decrease in size via breakage at the end of the fiber that results in monomer loss and is regulated by a first-order breakage rate constant, bS1 (units of s–1). (C) Fiber joining and breakage—fiber growth may also occur between any two fibers at a rate determined by the second-order rate constant, fJ (units of M–1s–1) and fiber internal breakage specified by a set of common site-specific intrinsic breakage rate constants, bS2 (units of s–1). End sites for breakage (characterized by breakage rate constant, bS1) are shown by a red arrow. Internal sites for breakage (characterized by breakage rate constant, bS2) are shown using a black arrow.
In developing a model of amyloid formation featuring position related breakage propensities, the following simplifying assumptions are made.
(i.) Amyloid growth follows a minimal mechanism (identical to that shown in Fig. 1), featuring fiber nucleation, fiber growth and dissolution via monomer addition/loss and fiber joining and breakage.
(ii.) The mechanism is characterized by a phenomenological fixed nucleus size set at 2 monomers along with specification of a single structural isomer type (per polymer degree) for all amyloid aggregates. Here, “phenomenological” refers to the fact that we do not seek to determine the correct ‘critical nucleus size’ but rather just specify forward and reverse nucleation rates which, through their variation, can empirically accommodate experimentally observed time courses of amyloid formation whilst keeping the nucleus size set at 2.
(iii.) There are two general first-order intrinsic breakage rates: bS1 (the end scission rate operating at terminal monomer bonds) and bS2 (the internal scission rate operating at internalized monomer to monomer bonds) (Fig. 1).
To further develop the model three averaged quantities are defined; (CE)TOT—the total number concentration of ends for amyloid growth assuming uni-directional growth [17,18] (Eq. 1—where z describes the degree of the longest amyloid polymer in solution at that particular time); (CP)TOT—the total concentration of monomer existing within amyloid extendable form (Eq. 2), and
| (1) |
| (2) |
| (3) |
Chemical rate equations describing the total concentration of amyloid ends and the total concentration of polymeric amyloid species can be specified as shown in Eq. 4 and 5 (with a derivation shown in Appendix 1).
| (4) |
| (5) |
Depending upon the nature of the simulated system the free monomer concentration, C1, may be considered as a constant [19] or else evaluated at each time point by difference between total protein and polymerized forms based on conservation of mass principles [19]. From inspection we note that Eq. 4 requires specification of the concentration of the phenomenological nucleus, C2. To provide a running time-dependent estimate for C2 (alongside the time-dependent estimates of (CE)TOT, (CP)TOT and
(i.) The amyloid number concentration distribution (at any particular time) is set to follow an exponential distribution defined by a time-dependent (and phenomenological) decay constant, k (Eq. 6) such that k≥0.
(ii.) The average polymer degree
(iii.) Once determined, the value of the dynamic decay constant, k, is used to provide an estimate of C2 upon insertion of the relevant quantities back into Eq. 6.
| (6) |
| (7) |
By adopting this process for the determination of C2, Eq. 4 and 5, together with Eq. 3, can be used to evaluate the time dependence of the formation of the total concentration of protein in the form of amyloid, (CP)TOT, the concentration of extendable ends, (CE)TOT, and the average polymer degree
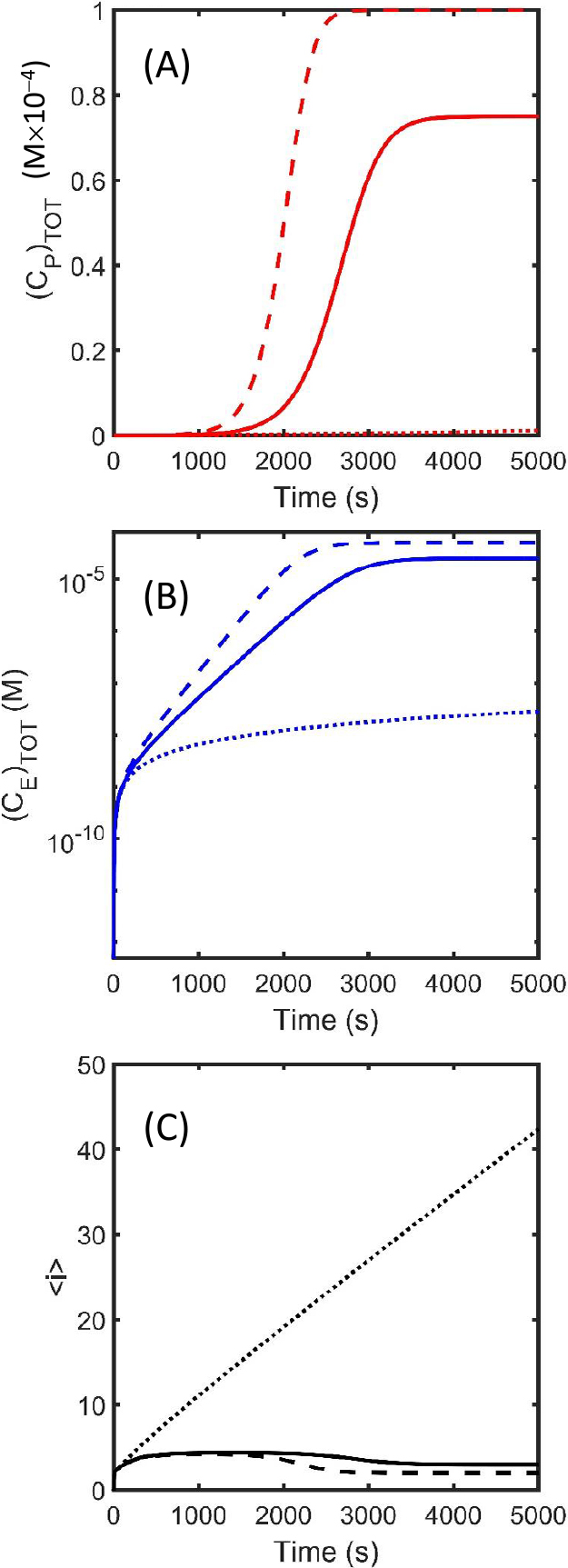
Effect on amyloid growth rate due to differences in internal (bS2) and end (bS1) breakage rates: Amyloid formation kinetics are simulated via numerical integration of Eq. 4 and 5 (utilizing the approximations derived in Eq. 6 and 7) for three relative cases reflecting a different ratio, θ, of the internal to end breakage rates such that θ=bS2/bS1. The simulations were carried out for a system where fiber joining does not occur using the following common values for additional parameter values, fN=0.001 M–1s–1, fG=200 M–1s–1, fJ=0 M–1s–1 and (C1)TOT=0.1 mM. The figure panels describe the formation of (A) (CP)TOT, (B) (CE)TOT and (C)
The described approach provides an extraordinarily simple means for incorporating differential fiber breakage susceptibilities occurring either internally, thereby creating two new fibers, or at the fiber ends, thereby releasing a monomer unable to maintain the amyloid structure. From Figure 2 we note that as θ increases from zero, the rate of total amyloid formation,
Although simulation results are dependent upon the values of the parameters adopted, confidence can be placed in the general trends shown in Figure 2 for the following three reasons. (i) Chosen parameters conform to a kinetically nucleated system (i.e. fG>>fN) thereby producing the classical sigmoidal kinetic curve that is a characteristic hallmark of amyloid growth [3–10]. (ii) Adopted parameters describe a thermodynamically nucleated system (helical polymerization) in which the total monomer concentration is much greater than the characteristic critical concentration i.e. (C1)TOT>>bS1/fG (when bS2=0) or (C1)TOT>>bS2/fG (when bS1=0) [1–4,19,21]. (iii) Simulations transition from one extreme limiting case of relative breakage to the other i.e. θ is examined over the range [0, ∞). In the present instance although different parameters will change absolute behavior (i.e. different time scale and extents) they will not change the relative behavior—the exposition of which is the chief interest in this Note.
The principle assumption within the paper involves the method for estimation of C2. To gain an appreciation as to why the adopted approach is valid we first note that the amyloid kinetic model is founded in the numerical integration of just two differential equations (Eq. 4 and 5) with the dependent variables describing the concentration of amyloid ends, (CE)TOT, and total monomer in amyloid, (CP)TOT (Eq.1 and 2). Due to its presence as a vanishing term in Eq. 4 and 5, error in the estimation of C2 will be virtually insignificant as the polymer distribution increases in size (i.e. C2/(CE)TOT→0; 2C2/(CP)TOT→0) thereby making the need for its accurate evaluation under these conditions unimportant [14,23]. At the other extreme, when C2 constitutes all of the amyloid fiber (i.e. C2/(CE)TOT=1; 2C2/(CP)TOT=1), its evaluation is exact (see Appendix 2). Within the intermediate region (as these fractions decrease from 1) the exponential assumption will essentially be a linear approximation, especially so for low average degree of polymerization e.g. [2 <
Two other notable assumptions made within the present work are (i) the use of a phenomenological nucleus of a size set at two monomers, and (ii) set equivalence of the nucleus dissociation rate and terminal monomer dissociation rate from the amyloid fibril. The first of these additional assumptions was made due to concerns based on partly practical and partly scientific reasons. The practical concerns are due to vagaries associated with classification of aggregate identity. If the nucleus size were to be appreciable (say 10 monomers) and formed in a stepwise fashion, then aggregate species between size 2 and 10 may be either amyloid or non-amyloid pre-nucleus in character. Determination of the nature of these species is experimentally difficult and further complicated by distinguishing whether or not they are associated with on, off or parallel pathways [14,26,27]. Scientific concerns relate to the way the nucleus size has been previously asserted via model based kinetic fitting. Results shown in Figure 2a clearly belie (or at least should raise a warning flag) any estimate of nucleus size based on use of fitting models featuring uniform breakage rates or size-independent growth rates [16,19,28]. As such, I would suggest that a minimal assumption of a nucleus size of 2 is suitable for the development of the current low parameter minimal model. Justification for the second of these additional assumptions (an assumed common rate of monomer dissociation from amyloid and nucleus i.e. both characterized by bS1) is harder to provide due to the fact that such information could only be supplied by isolated time-dependent observations made at the nanometer level of resolution (at present impossible). However, following an Occam’s razor type argument (in which models involving less parameters have greater scientific utility until proven inadequate) a judgement on this question should only be made on the basis of data analysis—for which no such suitable data currently exist.
Due to the fact that the method developed here does not describe an exact geometric dependence the current approach is likely applicable to the description of other systems involving one, two and three-dimensional polymerization processes. Like the model introduced by Smith, J. F., et al. [15] the current set of equations are simple enough that it can be easily incorporated into more complex simulation routines describing amyloidosis disease models [19,29,30], complex mixture models [14,26,31], signal identification and processing [13,27,32–34], polymer statistics based approaches involving amyloid formation from the unfolded protein state [8,28,35], or in ‘meta-models’ describing the transmission of prions in larger scale, more complex studies, involving organism level analysis of yeast, worms and flies [36–38]. Indeed use of Eq. 2 may help to cast light on the molecular mechanisms of candidates identified in ‘Anti-Prion’ screens [39].
I would like to thank (the now) Dr. Jeffrey Smith for a number of interesting discussions related to the present topic conducted during the course of his (JS’s) PhD. I acknowledge thoughtful comments from the two Reviewers who provided an exceptionally high quality review of the manuscript. I thank Dr. H.K. Edskes for helpful discussion and a highly enjoyable 15 years of collaborative work. I thank the Institute for Protein Research at Osaka University for the support they have provided over the last 5 years. I gratefully acknowledge the US Government for funds provided in the form of an ORISE Established Scientist Position. This research was supported in part by an appointment to the National Library of Medicine (NLM) Research Participation Program. This program is administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy (DOE) and the National Library of Medicine (NLM). ORISE is managed by ORAU under DOE contract number DE-SC0014664. All opinions expressed in this paper are the author’s and do not necessarily reflect the policies and views of NLM, DOE, or ORAU/ORISE.
The author declares no conflict of interest.
D.H. developed the theory and wrote the manuscript.
To derive Eq. 4 we begin by writing out the rate equation that corresponds to the actual set of allowable elementary steps.
| (A1) |
By noting (i) when i=2, bS2 C2+bS2 Ci(i–3)=0, and (ii) when i=3, bS2 Ci(i–3)=0, we can rewrite Eq. A1 as A2.
| (A2) |
Based on Eq. 1 and 2 we can rewrite Eq. A2 as Eq. 4.
With regard to Eq. 5 we similarly begin by writing out rate equation from allowable elementary steps (Eq. A3).
| (A3) |
Substitution of the identities shown in Eq. 1 allow Eq. A3 to be re-expressed as Eq. 5.
Appendix 2: Derivation of an analytical approximant for C2To obtain Eq. 7 we approximate the two discrete summations in the left-hand side of Eq. A4 with two separate integrals.
| (A4) |
With the value of