Review Article (Invited)
Why we are made of proteins and nucleic acids: Structural biology views on extraterrestrial life

キーワード:
origin of life,
astrobiology
2023 年 20 巻 2 号 論文ID: e200026

詳細
2023 年 20 巻 2 号 論文ID: e200026
Is it a miracle that life exists on the Earth, or is it a common phenomenon in the universe? If extraterrestrial organisms exist, what are they like? To answer these questions, we must understand what kinds of molecules could evolve into life, or in other words, what properties are generally required to perform biological functions and store genetic information. This review summarizes recent findings on simple ancestral proteins, outlines the basic knowledge in textbooks, and discusses the generally required properties for biological molecules from structural biology viewpoints (e.g., restriction of shapes, and types of intra- and intermolecular interactions), leading to the conclusion that proteins and nucleic acids are at least one of the simplest (and perhaps very common) forms of catalytic and genetic biopolymers in the universe. This review article is an extended version of the Japanese article, On the Origin of Life: Coevolution between RNA and Peptide, published in SEIBUTSU BUTSURI Vol. 61, p. 232–235 (2021).
One of the most important goals of the current space exploration missions is to find another life form (or its vestiges). What molecules should we look for to achieve the purpose? Can we expect extraterrestrial lives also have proteins and nucleic acids? Should we take completely different molecules into consideration? In this review, the essentially required properties for biomolecules (on the Earth and other planets) are discussed from structural biology viewpoints.
If a complex organism (as complex as the simplest bacterium we know on the Earth) were to emerge on another planet, then molecules to store genetic information and perform various chemical and physical functions must also have evolved. On the Earth, nucleic acids serve as information storage, and proteins play the main catalytic and mechanical roles. As no other examples of life have been discovered yet at the time of writing, we do not know if the terrestrial system (the combination of nucleic acids and proteins) is the only style in the universe or if there are totally different biomolecular systems. Still, we might be able to estimate the generally required properties for biological molecules in the universe, by carefully inspecting our own system. Constraints in the structures of biomolecules would be exceptionally pivotal, as their functions are fundamentally realized by their shapes and surfaces. In this review, the previous experiments to reconstruct ancient simple proteins are summarized, and then the properties of ideal biomolecules in the universe are discussed from structure biology viewpoints.
Numerous catalytic molecules are required for cellular survival. If the emergences of such molecules are miraculously rare, then we would be alone in this universe. However, if they are rather common events, then our universe might be full of life. Modern proteins on the Earth have overwhelmingly complicated sequences and structures, suggesting the difficulty of emerging by chance from non-living chemistry. This section outlines the experiments to demonstrate that folded proteins could have emerged in a plausible step-by-step evolutionary process from simple prebiotic peptides.
Secondary Structure AssembliesPeptides with secondary structures might have been intermediates between randomly synthesized prebiotic peptides and folded proteins [1–3]. For example, in 1975, Brack and Orgel reported that a simple peptide with alternating valine and lysine residues can self-assemble into large β-sheet structures [4]. The β-sheet structures were likely formed by simply orienting the lysine residues on one side to form a hydrophilic surface, and the valine residues on the other side to form a hydrophobic surface, which was likely further utilized to bind the hydrophobic sides of other β-sheets (Figure 1A). α-Helical assemblies were also formed by simple peptides with periodic hydrophobic patterns, which define the interaction surfaces with other peptide molecules (Figure 1B) [5]. Thus, primitive secondary structures and their assemblies can be formed by very simple sequences [6]. Furthermore, such peptides might have propagated by self-ligation reactions in peptide assemblies and become enriched on the ancient Earth [7–11].

Schematic structures of simple peptide assemblies with secondary structures. Hydrophilic and hydrophobic parts are colored blue and orange, respectively.
Peptide assemblies with secondary structures might also have played essential roles in co-evolution with RNA [1–3,12]. Our group has recently demonstrated that a peptide with hydrophobic and cationic moieties (P43: AKKVWIIMGGS) can form insoluble assemblies containing β-amyloid structures that accumulate RNA on their surfaces [13]. Indeed, longer RNAs bound more stably on the P43 aggregates even in high salt conditions. Furthermore, such peptides might also have supported RNA synthesis. RNA polymerase ribozyme (RPR) is an artificial RNA enzyme with an RNA-dependent RNA polymerase activity and regarded as a model molecule for the self-replicating RNA on the primordial Earth [14,15]. The aggregates of P43 and its sequence-simplified versions (e.g., K2V6: KKVVVVVV) enhanced the activity of RPR on their surfaces, although their effects were largely dependent on the buffer conditions [13]. Such β-amyloid peptides might have worked as selection platforms to concentrate longer RNAs from environments and support the functions of primordial ribozymes (Figure 1C).
Ancient Proteins with Smaller Sequence SpacesThere still seems to be a huge gap between simple peptides with secondary structures and modern proteins with tertiary structures. As modern proteins are polymers composed of 20 different amino acids, even a relatively small protein with 100 amino acid residues would have 20100 (~10130) possible sequences. It is almost unthinkable that functional sequences were efficiently searched from such a huge sequence space on the prebiotic Earth. In the early evolutionary stages of life, proteins might have emerged from a much simpler sequence library. For example, if an ancient protein could have emerged as a short peptide with 20 residues and five different amino acid types, the required sequence space would be only 520 (~1014), and its total mass could be less than 1 μg. To demonstrate that proteins with defined tertiary structures and functions can emerge from such smaller sequence spaces, the reconstructions of ancestral proteins with 1) shorter peptides and 2) fewer amino acid types have been performed, as described below.
The structures of some primitive proteins were probably created by the self-assembly of shorter peptides (10–50 amino acids) [16–19]. For example, the protein folds shown in the top panels of Figure 2 have internal pseudo-symmetries [20–25]. They are assumed to be descendants of ancient homo-oligomeric peptides, which evolved into monomeric proteins by gene fusion and then gradually lost the perfect symmetry by mutations in each repeating unit. Applying various protein engineering techniques, these pseudo-symmetric folds have been reconstructed as short homo-oligomeric peptides, or polypeptides with identical repeats (Figure 2, bottom) [26–37].

Structures of natural pseudo-symmetric proteins (A–D) and engineered symmetric proteins (E–H). Different monomers in oligomeric designs are colored with different colors. Multiple repeats in the monomer units are colored with different brightness.
Attempts to create proteins using fewer amino acid types have also been reported, resulting in the exclusion of 7 to 13 amino acid types for several proteins [36–45]. For example, an ancestral nucleoside diphosphate kinase (NDK), designed by using only 13 amino acids, maintained high thermal stability and enzymatic activity [44,45]. Even a further engineered NDK variant with only ten amino acid types could fold into the proper tertiary structure, although the activity was lost [44,45].
In a few cases, reconstructions by short peptides and smaller amino acid repertories were simultaneously achieved. For example, the β-trefoil fold was reconstructed as a polypeptide with three 42-amino-acid repeats containing only 12 amino acid types (Figure 2G) [36]. Another example is the DPBB fold conserved in various proteins, including the catalytic domains of cellular RNA polymerases. It was reconstructed by the homodimerization of a 43-amino-acid peptide containing only seven amino acid types (GAVDEK and R) (Figure 2H) [37]. Interestingly these seven amino acid types can be coded by GNN and ARR (R=A or G) in the modern genetic code. The five amino acids coded by GNN (GAVDE) are simple prebiotic amino acids. The other two (K and R) are positively charged and essential in interactions with nucleic acids. Thus, this might reflect an ancient amino acid repertory encoded by a primitive genetic code.
Structure Formation with Simplified Hydrophobic CoresThe formation of hydrophobic cores is essential for protein folding. Although simple β-amyloids or α-helical assemblies can be formed relatively easily [1–6], the formation of tertiary structures seems to be much more complicated. For protein stability, it is generally considered important to arrange the hydrophobic side chains without gaps in the hydrophobic cores. However, precisely packing multiple amino acid residues within the cores in various shapes would be exceptionally difficult for prebiotic chemistry or a primitive translation system with a lot of errors. To solve this problem, efforts to reconstruct folded proteins with simplified hydrophobic cores have been performed.
In the early days of protein engineering experiments, the hydrophobic cores of some proteins could reportedly be simplified by enriching one or two amino acid types [38]. A helix bundle protein (Rop) was reconstructed with a simplified hydrophobic core containing only alanine and leucine residues placed in a zigzag pattern (Figure 3A) [46,47]. Hydrophobic cores with more complicated folds (e.g., SrcSH3, T4 lysozyme, phage 434 Cro repressor) could also be extensively replaced by single amino acid types with relatively large structures (leucine, isoleucine, or methionine) (Figure 3B–D) [39,48–50]. These hydrophobic side chains have many possible rotamers (side chain conformations) and would fit within various shapes to fill the hydrophobic cores without gaps.

Engineered proteins with simplified hydrophobic cores. Experimental or predicted structures of engineered proteins with simplified hydrophobic cores are shown with their amino acid compositions. Protein sequences are adopted from each reference or PDB. Structural models of Rop Ala2Leu2-8, SrcSH3 FP2, and 434 Cro M-5 were predicted by Alphafold2 [51–53]. Structures of Rop Ala2Leu2-8 and mk2h_∆MILPYS are shown as homodimers. The C-terminal tag was removed from the model and amino acid composition of R2x2_VAL88. Hydrophobic side chains are shown as stick models. Alanine residues are colored yellow. Another enriched hydrophobic amino acid in each design is colored orange. Other hydrophobic residues and non-hydrophobic residues are colored black and light cyan, respectively.
However, the enrichment of such flexible side chains, especially the long linear one (methionine), would also be disadvantageous for protein stability, as they would cause large entropic losses when fixed inside hydrophobic cores [49]. This is probably the reason why long non-branched hydrophobic amino acids, like nor-valine and nor-leucine, were excluded from the proteinaceous amino acid repertory, despite their availabilities on the ancient Earth [54,55].
Recent examples of ancestral and de novo protein designs demonstrated that it is even unnecessary to fill hydrophobic cores with large side chains. The ancient DPBB fold mentioned above contains only the two shortest hydrophobic amino acid types (alanine and valine) (Figure 2H, Figure 3E) [37]. Koga’s group reported a very thermostable de novo protein with a hydrophobic core mostly composed of valine residues (Tm=106°C, Figure 3F) [56]. The structures of these ancestral and de novo proteins contain significant unfilled volumes within their hydrophobic cores. Interestingly, their overall structures were not compacted, as compared to the non-simplified variants with larger hydrophobic residues in the cores. These results indicated that the hydrophobic cores of some proteins can be formed without neat optimization, and even without large side chain fittings (by Leu, Ile, or Met) or zigzag patterning (Ala/Leu). In such cases, the formation and stability of the tertiary structures are likely to be more dependent on the designs of stable main chain structures (e.g., patterns of secondary structures and loops) [56–59].
Ancient protein evolution might have started with main chain topologies or folds that were stable even without neatly packed cores. Hydrophobic cores enriched with short side chains could have been more realistic, as simpler amino acids were probably plentiful on the ancient Earth. Furthermore, the presence of unfilled spaces in such cores would have been advantageous, as they could easily tolerate almost unavoidable contaminations with different amino acids by prebiotic chemistry or primitive translation systems. Such adaptability for random or bulky mutations is even seen in modern proteins [60,61].
Evolution from Simple Peptides to Folded ProteinsSimple peptides are considered to have existed on the prebiotic Earth [62]. For example, lysine-rich peptides and their analogs could have been synthesized by prebiotic chemistry [63–66], and mutually stabilized with nucleic acids [67]. As mentioned above, simple KV-rich peptides can form β-amyloid assemblies (Figure 1A) [4], capture RNA molecules, and support RNA synthesis by a model ribozyme (Figure 1C) [13]. These results suggest that such simple prebiotic peptides could have been sufficiently functional to support an RNA-based primitive life system on the ancient Earth.
Such peptides might have been conjugated and evolved into ancient (poly-) nucleotide-binding motifs with a few secondary structures [68–70]. P-loop and Helix-hairpin-Helix (HhH) are examples of such ancient motifs. P-loop and its flanking secondary structures have been suggested to be the evolutionary seed of a wide variety of nucleotide-binding enzymes (Figure 4A) [71–73]. Such peptides containing the β-(P-loop)-α element reportedly showed rudimentary helicase and adenylate-kinase activities [74,75]. HhH is a well-conserved nucleic-acid binding motif that adopts a pseudo-dimeric structure (Figure 4B), and a reconstructed ancient HhH motif can form a homodimer and assemble into macromolecular coacervates with RNA [76,77]. Furthermore, the DPBB fold conserved in the active site of modern RNA polymerase might have emerged by fusion between ancient RNA-binding peptides. The sequence of the ancestral DPBB design with seven amino acid types is highly KV-rich (eight lysine and fourteen valine residues in the 43 a.a. peptide) (Figure 2H, Figure 3E). Perhaps there were only a few steps between KV-rich RNA-binding β-amyloid peptides and β-barrel proteins [13,37].
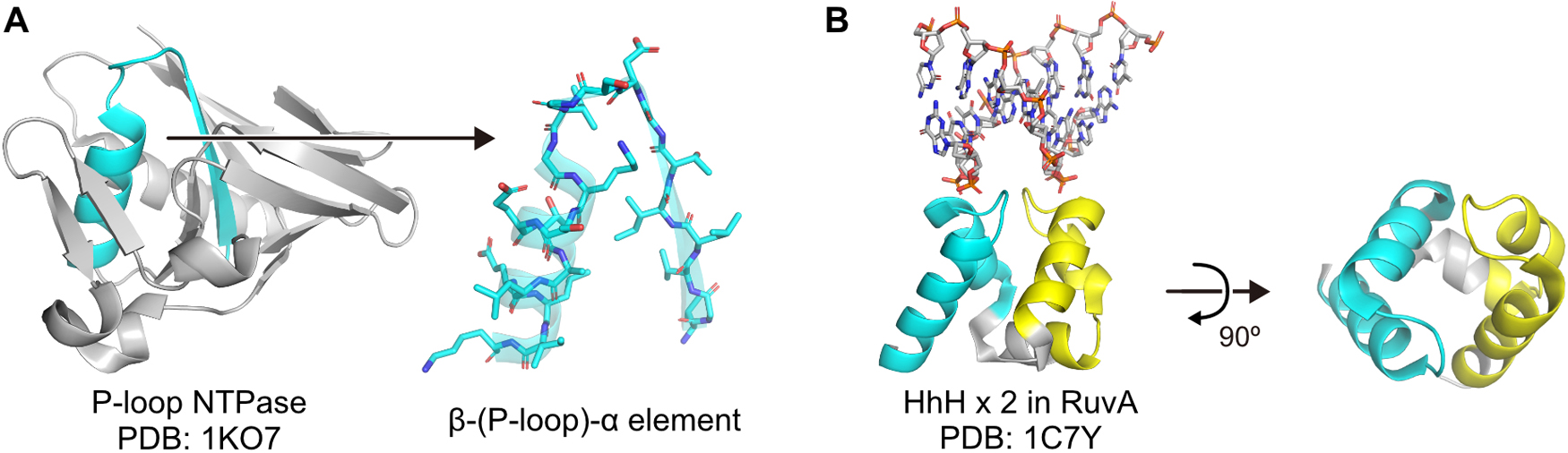
Structures of ancient (poly-) nucleotide-binding motifs. (A) Structure of the P-loop NTPase domain from HPr kinase and the closeup view of the β-(P-loop)-α element. The β-(P-loop)-α element is colored cyan. (B) Structure of the pseudo-dimeric HhH motifs in a Holliday junction binding protein, RuvA. The two HhH motifs are colored cyan and yellow, respectively.
Koga and colleagues successfully designed various protein folds by first linking secondary structure fragments with ideal loops for each topology, and then optimizing the side chains [56–59]. Similar processes might have happened in the early evolution of folded proteins. Protein folds with simple hydrophobic cores might have first emerged by fusion events between short peptides without sequence optimization, as wide range of sequences could be tolerated in reconstruction of simple protein folds [37,78]. As hydrophobic interactions do not have specificity or directionality, “hydrophobicity is nearly a sufficient criterion for the construction of a functional core”, which “would greatly reduce the initial hurdle on the evolutionary pathway to novel enzymes” [60]. Thus, the emergence of complicated protein structures might have been facilitated by relatively simple step-by-step procedures.
In contrast, nearly half of the amino acids in the modern genetic code are hydrophobic (8 out of 20 a.a. types), indicating the importance of their diversity. In very early protein evolution, a primitive genetic code might have encoded prebiotic hydrophobic amino acids (Ala, Val, Leu, Ile) without precise discrimination, as primitive hydrophobic cores could have formed by incorporating them almost randomly [37,78]. Then, through survival competitions, these amino acids might have been assigned to different codons, increasing the structural accuracies (and thus stabilities and activities) of the encoded proteins. Finally, paying higher synthetic costs, the genetic code might have started to incorporate larger hydrophobic amino acids (Met, Phe, Tyr, Trp). Such side chains likely have stronger aggregation tendencies [79], but also function, for example, in protein folding in lipid bilayers [80].
Now, the emergence of proteins on Earth seems not to have been so miraculous (even if it still had been a very rare event). What kinds of molecules would then be selected as counterparts of proteins and nucleic acids if a life system emerged and survived on another planet (or moon) somewhere in the universe? Would they be something (very) similar to ours, or totally different? In this section, I scrutinize the generally required properties of biological molecules responsible for chemical/mechanical functions and information storage, from a structural biology perspective.
Molecular Bonds and InteractionsFirst, the properties of representative molecular bonds and interactions are summarized in Figure 5, as they are the basis of the discussion in the following subsections (If you are confident about your textbook knowledge on this topic, you may skip this subsection. If you would prefer more detailed explanations, the recommended textbook is listed in the references [81]).

Molecular bonds and interactions. (A) Types of molecular bonds and interactions. (B) Properties of representative bonds and interactions in biomolecules.
All bonds and interactions inside or between biomolecules are fundamentally caused by electromagnetic forces. They are categorized into different groups depending on their apparent mechanisms (e.g., quantum mechanical, electrostatic, entropic) and properties (e.g., strength, specificity, directionality). Although such categorizations are somewhat ambiguous and occasionally cause confusion, they are still beneficial for a quick understanding of the nature of molecular bonds and interactions.
Covalent bonds have quantum mechanical properties, are formed by chemical reactions, and result in the synthesis of new molecules connected by shared electron orbits. Contrarily, other interactions result in physical contact between molecules without electron sharing. Electrostatic interactions involving charged atoms are further categorized into ion-ion and ion-dipole interactions. Van der Waals interactions are partially electrostatic and partially quantum mechanical, and contain three components: orientation (dipole-dipole), induction (dipole-induced dipole), and dispersion (induced dipole-induced dipole). While the interactions between ions and permanent dipoles can be attractive or repulsive depending on the involved molecules, the dispersion force is ubiquitous and always attractive. Although the dispersion force is usually weak, it can become stronger when electrons are only weakly caught in their orbits and easily disturbed by surrounding electrostatic fields (e.g., in large atoms and π bonds).
The hydrogen bond is roughly a special case of dipole-dipole interactions where an almost naked proton bridges two electronegative atoms (donor and acceptor) in a straight line. Because of the very small radius of the proton and large dipoles of the donor and acceptor molecules, the attracting Coulomb forces between the atoms are much stronger than in usual dipole-dipole interactions. Water has a tetrahedral molecular structure and forms three-dimensional networks of hydrogen bonds. To accommodate non-polar molecules in water, enough cavity spaces need to be formed against such strong hydrogen bond networks. Reorientations of water molecules surrounding non-polar molecules are also induced when they are solubilized in water, resulting in large entropy losses. Thus, non-polar molecules aggregate together in water to minimize the total non-polar surface and free as many water molecules as possible (hydrophobic effect). Although the resulting structures of non-polar complexes seem to be assembled by the non-polar dispersion force, it cannot be the driving force of the hydrophobic effect because the dispersion forces between non-polar molecules and between non-polar and water molecules are not very different. Thus, the hydrophobic interaction is mainly entropic and thus a long-range force, which can drive molecular assembly and folding in polar environments.
Figure 5B summarizes the properties of four representative bonds/interactions in biomolecules, especially focusing on pairing specificity and directionality (angle constraints). Covalent bonds can be formed between relatively free combinations of atoms. However, their spontaneous formations in biomolecules are observed only in rare cases (e.g., disulfide bonds between two cysteine residues), since they usually require specific catalytic reactions. Covalent bonds also have very strong directionality. Furthermore, double bonds have no rotational freedom and confer structural rigidity. Ion bonds require specific pairs (positive and negative) to be formed and have no directionality. Hydrogen bonds require specific pairs (donor and acceptor) to be formed and have relatively strong directionality. No other interactions can simultaneously have such pairing specificity and directionality, which confer the unique discriminating ability to hydrogen bonds. Contrarily, hydrophobic interactions do not require any pairing specificity or directionality. They just form between non-polar molecules in polar solvents. I will discuss which of these interactions would be ideal to realize the specific properties of ideal biological molecules, especially for various chemical/mechanical functions and information storage, in the following subsections.
Molecules for Chemical and Mechanical Functions: Most Likely ProteinsIf we suppose that some complex organisms (as complex as the last universal common ancestor, LUCA, on the Earth) could emerge on a planet, then numerous functional molecules would be required (like a few hundred proteins coded by LUCA) (Figure 6 (i)) [82]. To make the synthesis of such a large variety of molecules feasible, they would be composed of polymers with more or less uniform main chains and variable side chains (Figure 6 (ii)). Such polymers can be synthesized by a single polymerization system, and still achieve the huge diversity of the products.

Required conditions for ideal functional biopolymers.
To materialize numerous chemical and mechanical functions, the polymers also need to adopt various structures, as such functions are fundamentally accomplished by the shapes and surfaces of molecules (Figure 6 (iii)). In particular, high structural diversity at very small scales (sub-angstrom to a few nanometers) is essential to perform various chemical reactions in many metabolic pathways. In addition, if the polymers had defined structures, then their chains must have folded around cores (Figure 6 (iv)). What interactions would then be used to form such cores?
If numerous different shapes and cores had formed, then their emergence must have been easy enough to happen frequently (Figure 6 (v)). From this point of view, interactions with strict rules like covalent/ion/hydrogen bonds are unsuitable. Covalent bonds would not form easily or fit in different shapes either. It would also be impossibly complicated to solve hundreds of different 3D puzzles in such small scales by placing multiple charged or polar side chains interacting in the right combinations and right angles. Accordingly, we need to choose a force that does not have pairing specificity or directionality. Although the dispersion force meets this requirement, it would not be strong enough to form a core in non-polar environments. Thus, the only choice that allows a large variety of structures without meticulous design would be hydrophobic interactions in polar environments (Figure 6 (vi, vii)). As we have seen for the terrestrial proteins, the hydrophobic cores can form without a lot of optimizations and produce a huge variety of protein folds (The difficulty of the polar core formation might also be indicated by the structures of transmembrane proteins. Even in the lipid bilayers where the hydrophobic effect is negligible, it is obvious that transmembrane proteins do not fold around polar cores, but still mainly use hydrophobic interfaces. Although the folding mechanism of transmembrane proteins is not as well understood as that of soluble proteins, it is likely supported by several mechanisms, such as interactions between cationic/aromatic residues with the phosphate head groups of lipid bilayers [80]).
However, if the polymers were completely hydrophobic, they would just form irregular aggregates in the polar environments. Thus, here are the apparently contradicting demands: freedom to allow variety and restriction to define structures (Figure 6 (viii)). Such structural restrictions should be introduced by interactions with stricter rules while keeping the easy formation mechanisms. Thus, polar interactions (ionic or hydrogen bonds) must also be introduced to fold the polymers into well-defined 3D structures (Figure 6 (ix)).
But how can polar elements become embedded in hydrophobic cores? They must be introduced in well-paired (canceled) forms (Figure 6 (x)). In the case of our functional polymers (i.e., proteins), the introduction of a single polar residue in the hydrophobic core usually destabilizes the protein structure. In contrast, we sometimes find a pair of charged residues (salt bridge) even in a hydrophobic core. However, such well-placed pairs of polar side chains would not frequently occur by random mutations in hydrophobic cores and could not be the primary mechanism to introduce structural regularity into biopolymers in the universe. Thus, the polar interactions in hydrophobic cores are much more likely to be achieved by the main chains with regular repeating units (Figure 6 (xi)).
To embed the main chains in hydrophobic structures, all polar parts in their repeating units should ideally be canceled by pairing with each other. In other words, the numbers of two pairing groups in the main chain should be the same (Figure 6 (xii)). For example, in one of the simplest cases, the main chain would have one hydrogen donor and one acceptor in its repeating unit, which would pair perfectly without leftovers.
An additional way to balance the freedom and regularity in the polymer structures might be the introduction of partial rigidity in the main chains (Figure 6 (xiii)). For example, double bonds or cyclic structures would restrict bond rotations and make the polymer tend to adopt regular conformations. If evolutionary competitions selected more suitable molecules, then the biopolymers of surviving organisms might also have main chains with such optimized rigidity.
Finally, even after fulfilling the above conditions, the monomer units of the biopolymers must be kept simple (Figure 6 (xiv)). They need to be synthesized by non-biological chemistry or by very primitive life systems. Also, not only diversity of sequence but also diversity per volume would be essential to realize various functions. If the monomer units were too large (e.g., 50 Å), then the polymer would never have optimum binding surfaces for multiple substrates.
Summing up the above arguments, functional biopolymers should fold into various shapes and have hydrophobic cores (Figure 6 (iii, vi)). They must have main chains with the same numbers of two groups to form polar interactions (Figure 6 (xii)), ideally with partially rigid structures (Figure 6 (xiii)). Their monomer unit should also be as small as possible (Figure 6 (xiv)). What molecules satisfy such requirements? Living on the Earth, we know one example, poly-α-amino acids (i.e., proteins) (Figure 6 (xv)).
The repeating unit of poly-α-amino acids has one donor and one acceptor of a hydrogen bond, which are used in extensive intramolecular interactions (self-complementary) [83]. Peptide bonds also have a double bond property because of their resonant structures. These bonds have an almost fixed plane, while still leaving partial rotational freedom around the α-carbon (φ and ψ angles) [84]. Thus, the requirements for ideal functional biopolymers are met by the six atoms in the peptide bond (Figure 6 (xv)). This also seems to be at least one of the simplest polymers to fulfill the above conditions. Considering that amino acids are quite common in the universe and peptide bonds can easily form even without life [62], we would expect some significant population (maybe the majority) of extraterrestrial lives to use proteins to perform chemical and mechanical functions. Therefore, the conclusion here is the same as the one from a seminal review by Weber and Miller: “If life were to arise on another planet, we would expect that the catalysts would be poly-alpha-amino-acids...” [54].
Molecules for Information Storage and Transfer: XNAWhat molecules would store the genetic information of extraterrestrial lives? The simplest way we know to save and convey information is a sequence of letters (Figure 7 (i)). Molecules for information storage would most likely materialize this concept. For this purpose, again, polymers with uniform main chains (sequencing) and different side chains (letters) would be the easiest choice (Figure 7 (ii)). What kind of interactions would they use? What kind of shapes would they adopt?

Required conditions for ideal genetic biopolymers.
Information precision and high distinctiveness would be the most important properties required for letters in genetic polymers (Figure 7 (iii)). Such letters would somehow be materialized by bonds or interactions with high specificities. Considering that genetic information should not only be stored but also transmitted, the letters are likely read and written through interactions that can be formed, dissociated, and reused easily. In the bonds and interactions listed in Figure 5B, the hydrogen bond is apparently the best candidate to meet these requirements, as it has pairing specificity, directionality, and can be formed by physical contact (Figure 7 (iv)).
In contrast to functional biopolymers with hydrophobic cores and various structures, the genetic polymers with hydrogen-bonded letters would have much more regular structures. As I argued above, placing multiple polar side chains in various 3D puzzles would be a highly complicated problem, since we cannot place them randomly, unlike the cases of non-polar side chains for hydrophobic interactions. Thus, only finite interaction patterns between limited components would arise. In other words, a few different “pairs” of side chains would work as letters in the genetic polymers (Figure 7 (v)).
It seems reasonable to assume that the structures of these few side chain pairs would be somewhat similar to each other, except for the discriminators of letters (hydrogen bonds), because they need to meet the same functional and structural requirements (also see below for a more detailed discussion about the structures of “base” pairs). Such more or less uniform side chain pairs must be connected by uniform main chains and repeated multiple times to encode genetic information (Figure 7 (vi)). If we connect and repeat a uniform structure in a uniform way (a linear, head-to-tail polymer), it would be a helix (Figure 7 (vii)) [85]. As a genetic polymer contains paired side chains, it would be a double helix (Figure 7 (viii)).
What structures, then, should the paired side chains have? To discriminate different letters, they should have at least a few hydrogen bonds. Larger numbers of the pairing bonds might increase the stability and specificity of the pairs, thus contributing to the precision of information storage and transfer. However, it would be more difficult to synthesize larger side chains, and finding the complementary pairs would also be challenging. For example, the probability for randomly ordered donors and acceptors from two side chains to form ten perfectly-coupled hydrogen bonds would be 0.1% (1/210). Realistically, the pairing side chains in genetic materials would only have a few hydrogen bonds per pair (Figure 7 (ix)).
However, this estimation raises another problem. In the discussion about functional biopolymers above, I estimated that such biopolymers (and life) would emerge in polar environments to allow various hydrophobic cores to form (Figure 6 (vii)). In such polar environments, polar interactions become weak (Figure 7 (x)). A few hydrogen bonds are not sufficient to stably couple side chains, as such polar regions can also form hydrogen bonds with surrounding water molecules. To overcome this problem and to guarantee stable pairing between letters, a few features must be present in the side chains: 1) rigidity, 2) hydrophobicity, and 3) flat conformation.
First, the discriminators of the letters (donors and acceptors of hydrogen bonds) should be fixed on rigid basement structures, or “bases” (Figure 7 (xi)). If the pairing side chains have flexible structures and the hydrogen bond donors and acceptors also fluctuate, then the chances of having the proper pairing would drastically decrease. The hydrogen bonding between fluctuating structures would also cause large entropic losses by fixing them in the paired conformations. If the pairing side chains have rigid bases to fix the hydrogen bond donors and acceptors, then such problems can be avoided.
Second, the polar discriminators of the genetic letters should ideally be sequestered in a hydrophobic layer to enhance the recognition abilities of hydrogen bonds (Figure 7 (xii)). Such hydrophobic parts would also be present in the side chain bases. As the surrounding environments are supposed to be polar, the genetic polymer should also have an outer hydrophilic layer. Thus, the round slice of the genetic polymer would have the hydrogen-bonded pairings at the center of the double helix, the side chain bases as the middle hydrophobic layer, and the outer hydrophilic layer likely formed by the main chain.
This ideal architecture of genetic polymers leads to an additional requirement for the structures of the side chain bases. As they are repeated and trapped in the inner space of the double helix, they must be piled up neatly. Thus, flat structures would be favored (Figure 7 (xiii)). If the bases of the side chains had bumpy structures (like the chair and boat conformations of cyclohexane), then there would be unfavorable unfilled spaces when stacked in a helical structure. In contrast, the neatly piled-up structure of the flat hydrophobic bases would contribute to the stability of the overall double-helical structure of the genetic polymer by interactions between stacking bases. These stacking interactions could be further enhanced if they contain strongly induced dipoles, for example, in π bonds.
Summing up the above arguments, the optimal side chains in genetic polymers would have rigid and flat base structures with hydrophobic moieties (ideally containing strongly induced dipoles). Fortunately, these conditions can be solved by a simple solution: double bonds with sp2 orbitals (Figure 7 (xiv)). Double bonds confer rigidity to the molecular structures by restricting the rotation around them. Sp2 contains three hybrid orbitals in a plane. The hydrogen bond donors and acceptors would also stick out in the same plane, supporting their interactions in the proper straight-line conformation. Furthermore, the π electrons in double bonds would have strongly induced dipoles and enhanced hydrophobic stacking between bases. If the ring structures are formed by multiple double bonds and sp2 orbitals, then they would be especially suitable, since they are very rigid, flat, and rich in π electrons.
Additionally, to support weak recognitions by a few hydrogen bonds, it would be more desirable if the pairing bases have size complementarity (i.e., the small one forms a pair with the large one), which would be a strong discriminator when trapped in the limited space inside the double helix (Figure 7 (xv)). Considering the parsimonious aspect of evolution, this size complementarity would be achieved by the minimum required structures: a one-ring base forms a pair with a two-ring base.
On the Earth, we know one example of side chains meeting the above conditions for the ideal genetic polymers: nucleobases (Figure 7 (xvi)). They pair by a few hydrogen bonds at the center of the double helix structures of DNA and RNA [86]. They also have rigid and flat ring structures with several double bonds (including resonance structures). They are piled up neatly inside the double helix, stack with each other by their rich π electrons to form the hydrophobic middle layer, and stabilize the paired helix [87–94]. The size complementarity between one- and two-ring structures is also present in nucleobases. Thus, although the existence of different letters with similarly ideal or exceptional properties cannot be excluded (e.g., a hydrophobic shape-complementary pair of artificial nucleobases) [95], the structures of the terrestrial genetic letters seem to be at least one of the simplest forms in the universe to meet such a number of conditions for ideal genetic letters.
In contrast to the side chains, various choices might be allowed for the main chains of the genetic polymers, as they are likely to be the outer layer and would have fewer structural restrictions. The main chains of the terrestrial genetic polymers are composed of sugars and phosphate. The sugars can be different even in our life system (ribose and deoxyribose), and can also alter the overall conformation of the nucleic acids (A-form and B-form). Artificial nucleic acids (or xeno nucleic acids, XNA) with different sugars (e.g., HNA) or even with non-sugar counterparts have also been developed and shown to store and convey information [96,97]. Thus, ribose and deoxyribose might have been selected by chemical availability or stability (including the stability of their double helix) under some specific conditions on the Earth.
Phosphate might also have been chosen for its chemical properties. It is not a structurally essential component as XNA without phosphate can also form base-paired double helices (e.g., PNA) [96–98]. With its negative charge, phosphate can repulse the attacking OH– in the aqueous environment, making the nucleic acids relatively stable [99]. Another chemical reason might be the strategy to achieve the synthesis of nucleic acids without uncontrolled degradation. Polymerization (dehydration) of monomer parts of nucleic acids (NMPs/dNMPs) does not occur spontaneously in aqueous environments. Thus, the substrates of the polymerase reactions must somehow become activated. Among a lot of possible activated substrates, polymerases do not use the simplest ones (NDPs/dNDPs), but the doubly activated ones (NTPs/dNTPs), paying higher synthetic costs. The reason for this choice is to prevent the reverse reaction [100,101]. After the incorporation of an NMP/dNMP in the elongating strand, pyrophosphate (PPi) is released and further degraded into two phosphates, which prevents PPi from re-entering the catalytic site of polymerases to trigger the reverse reaction (pyrophosphorolysis). If polymerases were to use NDPs/dNDPs as substrates, then the leaving group of the polymerization reaction would be phosphate, which would not be further degraded and inevitably cause the reverse reaction.
The doubly activated substrates can also function in another chemical equilibration (NTP↔NDP+Pi) in the same environment. Cells likely utilize this for reversible reactions that should be controlled in temporal energy/nutrition richness. Using the same substrates, non-reversible reactions including genetic polymer synthesis and reversible reactions in response to the fluctuating environments can be simultaneously performed with different chemical equilibrations [101]. The few repeating phosphates in a nucleotide are likely one of the simplest ways to realize such an elaborate control system for the complicated traffic of numerous chemical reactions in the cell. Furthermore, lipid membranes assembled by hydrophobic interactions can prevent phosphorylated cellular ingredients (e.g., nucleic acids and numerous metabolic intermediates) from leaking out, keeping the integrity of the cell [99,102]. If such phosphate chemistry systems were also adapted on another planet, then their genetic polymer might be something similar to ours.
Another possible reason for the main chain choice of nucleic acids on the Earth might have been related to the enzymatic functions of RNA. Most natural ribozymes perform phosphoryl transfer reactions, such as self-cleavage and its reverse ligation [103]. The mechanism is essentially an SN2-type nucleophilic attack on the phosphodiester bond by a 2′ oxygen or a hydroxide ion, and sometimes catalyzed by Mg2+ ions chelated between nearby phosphate groups. In other words, the RNA structure seems to be optimized for such self-editing reactions, while proteins catalyze diverse metabolic reactions. This might indicate RNA originally emerged as a selfish self-replicator [104,105].
In this review, I have discussed the structures of ideal biopolymers for elaborate life systems, considering the properties of representative bonds and interactions. This process was almost like reading a biochemistry textbook in reverse and discovering how our biopolymers elegantly meet the required conditions.
To perform their numerous chemical and mechanical functions, functional biopolymers must fold around hydrophobic cores and adopt various structures. They also need to have relatively rigid main chains with one hydrogen bond donor and one acceptor, to introduce regularity into otherwise random hydrophobic structures. Proteins are probably the simplest molecules to fulfill such conditions. They might also be the functional polymers of many extraterrestrial lives.
In contrast, genetic polymers should store and transmit information precisely. They need to form double helices with flat, stacking side chains paired by a few hydrogen bonds. DNA and RNA have ideal side chains, the nucleobases. Their main chains might have been selected for more chemistry-based reasons. The genetic polymers of extraterrestrial life might have some variety, especially in their main chains. Still, some of them might be very similar or even the same as ours.
It is worth mentioning that all of the above arguments suppose the existence of highly evolved lives. Simpler life-like entities composed of non-ideal molecules might have been present before the emergence of more elaborate life systems, or in special environments. Even on the Earth, nucleic acids or peptides might have played both catalytic and genetic roles in very primitive life systems, without cooperating with each other [1,104,105]. For instance, nucleic acids can catalyze chemical reactions (sometimes using modified nucleobases) [106–108], and peptides might also undergo self-templated replication [7–11].
However, ideal genetic polymers like DNA and RNA cannot be as functional as proteins, as they are all thumbs. Their side chains are inevitably large and have limited varieties, which would result in much less diversity per volume. In contrast, ideal catalytic polymers like proteins have various side chains, including small hydrophobic ones, which would not be replicated precisely. Therefore, the co-evolution between the ideal genetic polymers and the ideal catalytic polymers would be highly advantageous (molecular mutualism) [109] and might also be the only way to generate complicated life systems like ours.
The author declares no conflicts of interest.
S.T. wrote the manuscript.
The evidence data generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
I was supported by JSPS (22H01346). I thank Sota Yagi for fruitful discussions.