Abstract
Optogenetic techniques offer a high spatiotemporal resolution to manipulate cellular activity. For instance, Channelrhodopsin-2 with global light illumination is the most widely used to control neuronal activity at the cellular level. However, the cellular scale is much larger than the diffraction limit of light (<1 μm) and does not fully exploit the features of the “high spatial resolution” of optogenetics. For instance, until recently, there were no optogenetic methods to induce synaptic plasticity at the level of single synapses. To address this, we developed an optogenetic tool named photoactivatable CaMKII (paCaMKII) by fusing a light-sensitive domain (LOV2) to CaMKIIα, which is a protein abundantly expressed in neurons of the cerebrum and hippocampus and essential for synaptic plasticity. Combining photoactivatable CaMKII with two-photon excitation, we successfully activated it in single spines, inducing synaptic plasticity (long-term potentiation) in hippocampal neurons. We refer to this method as “Local Optogenetics”, which involves the local activation of molecules and measurement of cellular responses. In this review, we will discuss the characteristics of LOV2, the recent development of its derivatives, and the development and application of paCaMKII.
Significance
Optogenetic techniques are valuable for the precise manipulation of cells in both space and time. Nevertheless, the absence of adequate optogenetic molecular tools makes it difficult to manipulate intracellular molecules in restricted regions like synapses. The best way for local manipulation is to combine 2-photon excitation and LOV2 since it is efficiently activated by 2-photon excitation. This review presents the optogenetic tools based on the LOV domain developed so far, which we hope will facilitate their comprehension and further advancement. Moreover, we will provide an example of “Local Optogenetics” involving paCaMKII and two-photon excitation.
Introduction
Optogenetic techniques have been extensively utilized in recent decades to manipulate cellular activity [1,2]. In optogenetics, light is combined with genetically-encoded photoactivatable proteins to control and manipulate the activity of specific cells in living organisms. Compared to conventional cell manipulation techniques such as drug treatment and gene down-regulation, optogenetics offers several advantages, including high spatiotemporal resolution, cell-type specificity, and reversibility. Opsins, including Channelrhodopsin-2 (ChR2), in combination with global light illumination, are the most widely used to control neuronal activity at the cellular level (Fig. 1A) [1,3]. However, the application of optogenetics to the subcellular level has been limited despite its importance. For instance, since individual synapses operate independently on a neuron, being able to manipulate them at the single-synapse level optically would be an extremely valuable technique in studying neuronal functions and memory (Fig. 1B). However, while current optogenetic tools primarily enable manipulation at the neuronal level (a scale of several tens of μm), manipulating small areas such as synapses (less than 1 μm) has not progressed due to the lack of appropriate optogenetic tools. For instance, although ChR2 can be locally activated by 2-photon excitation, the induction of synaptic plasticity at the level of single synapses has not been achieved [4–6].

The Light-oxygen-voltage (LOV) domain 2 (LOV2) of Phototropin 1, a plant photoreceptor protein, is a soluble protein in the cytosol (Fig. 2A). It has a higher two-photon absorption efficiency than other photoresponsive molecules such as Cryptochrome 2 (Cry2), making it ideal for two-photon excitation [7–9]. The creation of LOV2 mutants with various properties has been widely explored, and the crystal structures of LOV2 are available, providing an opportunity for developing novel optogenetic tools. Thus far, a wide range of optogenetic tools based on LOV2 have been created, enabling the induction of various biological events such as transcription and cell shape changes through light (Table 1). Recently, we engineered a photoactivatable CaMKII by fusing LOV2 with Ca2+/calmodulin-dependent protein kinase II (CaMKII) [10]. This new tool can be activated by blue light (400–500 nm) or near-infrared (700–980 nm) two-photon excitation. By activating paCaMKII in excitatory neurons by two-photon excitation, synaptic plasticity can be induced at the single synapse level, allowing to study the functions of CaMKII and synaptic plasticity [10,11]. This review will discuss the features of LOV2 domains, recent advances in their mutants, and the development and applications of paCaMKII as an extended version of our Japanese article [12].

Table 1
LOV2-based optogenetic tools
|
Mechanisms of action |
Example applications |
| LOV2 |
Conformational change (undocking of Ja helix from PAS core) |
GTPase signaling [17,20], ion channel [21,22], intrabody regulation [82,83], CaMKII regulation [10,84], kinase inhibitor [85–87], protein degradation [88,89], nuclear transfer or export [23–25], peroxisome translocation [90], myosin and kinesin motors [91], condition-dependent transcriptional regulation [92–95], Cre recombinase [96], Cas9 regulation [97,98], Endonucleases [99], RNA polymerase [100], apoptosis [101], membrane binding [102] |
| TULIP |
Heterodimerization between LOVpep and ePDZ |
organelle transport [103], GTPase signaling [104,105] |
| iLID |
Heterodimerization between iLID (SsrA) and SspB |
GTPase signaling [106–108], microtubule dynamics [109], ion channel [110], inter-organelle contacts [111], phase separation [112,113], mitophagy [114], actomyosin contractility [115], organelle transport [116] |
| LOVTRAP |
Dissociation between LOV2 and Zdk |
microtubule dynamics [117], transcriptional regulation [89], GTPase signaling [118,119], steric inhibition of protein active sites [120] |
| AtLOV2 |
Conformational change (undocking of Ja helix from PAS core) |
protein degradation [31] |
| BcLOV4 |
The membrane binding region masked by RGS domain is exposed and binds to the membrane |
GTPase signaling [34,121,122] |
| EL222 |
Homodimerization of DNA binding domain |
transcriptional regulation [35,123] |
| FKF1 |
Heterodimerization between FKF1 and GIGANTE |
GTPase signaling [36], transcriptional regulation [36,124], ion channel [125] |
| VfAU1 |
Homodimerization |
RTK signaling [37,126,127], Nodal signaling [128], ion channel [129] |
| RsLOV |
Dissociation |
transcriptional regulation [130], Cas9 regulation [39] |
| YtvA |
Conformational change in the homodimer (rotation of each unit) |
transcriptional regulation [131–133], Histidine kinases [41], Gac/Rsm signaling [134] |
| VVD |
Homodimerization and Heterodimerization between VVD and NcWC1 |
transcriptional regulation [42,135], RNA regulation [136], Cre recombinase [137,138], organelle transport [116], Tyrosine kinases [139], Zn2+ binding [140] |
| Magnets |
Heterodimerization between positive Magnet and negative Magnet |
transcriptional regulation [141], RNA polymerases [142], Cre recombinase [143,144], Cas9 regulation [145,146], inter-organelle contacts [147], cell-cell adhesions [148], intrabody regulation [149] |
| NcWC1 |
Heterodimerization between VVD and NcWC1 |
transcriptional regulation [44] |
Characteristics of the LOV2 Domain and its Variants
The LOV domain is a photosensory domain that absorbs blue light [13], and it has been identified in bacterial, algal, fungal, and plant species. The LOV2 domain, which is derived from Avena sativa Phototropin 1 (AsLOV2), is a representative LOV domain [14]. It consists of 143 amino acids (404–546 a.a. in Phototropin 1), and its C-terminal Jα helix tightly binds to the Period-ARNT-Singleminded (PAS) core of LOV2 in the dark state (Fig. 2A). When LOV2 is exposed to blue light, flavin mononucleotide (FMN) embedded in LOV2 is excited and forms a covalent bond with Cys450 (Fig. 2A). This covalent bond induces a conformational change in LOV2, causing the release of the Jα helix from the PAS core of LOV2 in a reversible manner, with a deactivation half-time of approximately 40 seconds in the dark state. Since the FMN is also excited by the broad wavelength range of 2-photon excitation, it should induce the structural change of LOV2 (Fig. 2B). The deactivation half-time can be adjusted to range from seconds to hours by introducing mutations in LOV2 [13,15,16]. Typically, when designing optogenetic tools based on LOV2, an active protein such as small GTPase Rac1 is C-terminally fused to Jα helix of LOV2, with linker optimization to inhibit or inactivate the active protein [17]. Upon blue light illumination, the Jα helix is released from LOV2, allowing the sterically hindered active protein to gain its native function. Recently, circularly permuted LOV2 (cpLOV2) has been developed. It allows the fusion of a protein to the N-terminal of LOV2 [18,19]. cpLOV2 is created by fusing the Jα helix (517–546 a.a. of LOV2) to the N-terminal of LOV2 (404–516 a.a.) with a flexible linker.
A variety of optogenetics tools based on LOV2 have been developed to manipulate GTPase signaling [17,20], ion channel [21,22], nuclear transfer signaling, nuclear export signaling [23–25], and many other uses (Table 1). Furthermore, LOV2-based translocation, oligomerization, and dissociation tools have been developed, including tunable light-inducible dimerization tags (TULIPs) [26], improved light-induced dimer (iLID) [27], and LOV2 trap and release of protein (LOVTRAP) [28]. TULIPs are composed of a modified PDZ domain (ePDZ) and LOVpep. LOVpep is a fusion of LOV2 and an ePDZ-binding peptide (pep). In the dark state, the pep fused to LOVpep cannot bind to ePDZ due to steric hindrance by the LOV2-Jα interaction. Upon blue light illumination, the Jα helix and pep dissociate, and the pep becomes exposed, allowing it to bind to ePDZ. iLID consists of LOV2-Jα-SsrA and SspB. Upon exposure to blue light, they form heterodimers similar to TULIPs. In contrast, LOVTRAP consists of LOV2 and its synthetic binding partner Zdark (Zdk), which recognizes the dark state of LOV2 and forms a heterodimer. Upon blue light, the structural change of LOV2 occurs, leading to the dissociation of Zdk from LOV2. These LOV2-based dimerization and dissociation systems have been widely employed in developing various optogenetic tools (Table 1).
Moreover, Arabidopsis thaliana-derived LOV2 (AtLOV2) shares a similar structure and amino acid sequence with LOV2, exhibiting 80% homology with a major difference in the linker region between LOV2 and Jα helix [30]. Using AtLOV2, a light-dependent degradation system was developed by fusing a proteasome recognition sequence, cODC1, with AtLOV2 [31].
The LOV domain derived from Botrytis cinerea (BcLOV4) can bind anionic phospholipids. Upon blue light illumination, the membrane-binding region in the LOV domain becomes exposed and subsequently binds to the cell membrane [32,33]. By fusing BcLOV4 with the small G protein RhoA and expressing it in HEK293T cells, local light stimulation successfully induces cell contraction by recruiting RhoA to the cell membrane [34]. One notable advantage of BcLOV4 is that it is possible to construct a light-dependent membrane translocation system using a single component.
The EL222 transcription factor, derived from Erythrobacter litoralis, comprises LOV and DNA-binding helix-turn-helix (HTH) domains. In its dark state, it remains monomeric; however, upon exposure to blue light, it forms a homodimer and binds to DNA [35]. The EL222 has been fused with the V16 transcriptional activation domain and co-expressed with the target gene possessing five consecutive EL222 binding sites (C120) upstream of the TATA box. Upon blue light illumination, EL222 binds to C120, activating the target gene’s transcription [35].
The LOV domain-containing flavin-binding Kelch repeat F-box1 (FKF1) forms heterodimers with the GIGANTEA protein (GI) upon blue light illumination [36]. One notable characteristic of FKF1 is its irreversibility. By fusing FKF1 with Rac1 and adding a CAAX motif to GI to localize it to the membrane, lamellipodia formation can be induced by recruiting Rac1 to the membrane upon blue light illumination [36].
The LOV domain of the transcription factor Aureochrome 1 derived from Vaucheria frigida (VfAU1) exists in a monomeric form in the dark and forms a homodimer upon blue light illumination [37]. One application of VfAU1 is the activation of receptor tyrosine kinase (RTK) signaling. By fusing VfAU1 to the C-terminal of the intracellular domain of a receptor tyrosine kinase FGFR1, which can be activated in a light-dependent manner [37].
The LOV domain of Rhodobacter sphaeroides (RsLOV) consists of an N-terminal Aα helix and C-terminal Jα and Kα helices. It forms a homodimer in the dark state and dissociates into monomers upon blue light illumination [38]. A tool that utilizes RsLOV to regulate Cas9 has been developed. The fusion of RsLOV and Cas9 forms a dimer in the dark state, inhibiting Cas9 activity. Upon blue light illumination, the dimer dissociates, restoring Cas9 activity [39].
YtvA, which contains a LOV domain derived from Bacillus subtilis, forms a homodimer in the dark state. Upon blue light illumination, each unit of the homodimer undergoes a 5-degree rotation, leading to a structural change [40]. One application of YtvA is for the control of histidine kinases. By replacing the N-terminal PAS domain of the histidine kinase FixL with YtvA, the kinase is activated in the dark state and inactivated upon blue light illumination [41].
The LOV domain of the Vivid protein (VVD) from Neurospora crassa forms a stable homodimer upon blue light illumination with a slow dissociation rate of several hours [42,43]. Furthermore, a modified variant called Magnets, which forms heterodimers between pMag (positive Magnet, I52R/M55R mutant) and nMag (negative Magnet, I52D/M55G mutant), has been developed [43]. An application of VVD is the regulation of light-dependent gene expression by combining it with the GAL4/UAS gene expression system [42]. By using Magnets, PI3 kinase can be recruited to the cell membrane to alter membrane morphology [43]. Similarly, the LOV domain of White Collar-1 from Neurospora crassa (NcWC1) forms a heterodimer with VVD upon blue light illumination [44] and is applied to the gene expression system [44].
Cryptochrome 2 (Cry2) is another type of blue light-responsive protein that differs from LOV2 but is used extensively. The N-terminal photolyase homology region (PHR) of Cry2 contains flavin adenine dinucleotide (FAD), and Cry2 forms heterodimers or homomultimers upon blue light illumination [45,46]. Heterodimer formation occurs between wild-type Cry2 and CIB1 [47,48]. Modification has made dimer formation between the PHR of Cry2 and the truncated C-terminal of CIB1 (CIBN) possible [48–50]. Homomultimer formation of Cry2 has been reported not only for the wild-type but also for improved versions such as Cry2olig and Cry2clust [51–53].
Spine Signaling and Structural Long-Term Potentiation
Long-term potentiation (LTP), a type of synaptic plasticity, refers to a long-lasting strengthening of synaptic transmission. The mechanism of LTP has been extensively studied in the excitatory synapses of the hippocampus [54]. First, presynaptic glutamates bind to N-methyl-D-aspartate (NMDA)-type glutamate receptors (NMDARs) on the surface of the dendritic spine, which is the small protrusion (<1 μm) emanating from the dendrite and contains the postsynapse (Fig. 1B). Second, Ca2+ influx is triggered through the glutamate-bound NMDARs [55]. This Ca2+ increase leads to the activation of various intracellular signaling molecules, including calmodulin [56,57], which binds to CaMKII [56–59]. As a result of the structural change in CaMKII, its kinase activity increases [60,61], which in turn phosphorylates and recruits signaling molecules [62–64]. These events cause spine enlargement and accumulation of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)-type glutamate receptors (AMPARs) in the postsynaptic density, which is known as LTP [65–67]. It is worth noting that previous studies have shown that the activation of CaMKII alone can induce LTP [10,68–70]. As prior research indicates that persistent spine enlargement corresponds with an increase in AMPA currents (i.e., LTP) [71–73], this spine enlargement is referred to as structural LTP (sLTP).
CaMKII
CaMKII is a crucial signaling protein for inducing LTP [56–59] and is abundant in cortical and hippocampal neurons (1–2% of total protein in neurons). CaMKII is a serine/threonine protein kinase consisting of 12–14 α and β subunits (50 and 60 kDa, respectively) at a 3:1 ratio [74,75]. Upon binding of Ca2+/calmodulin to the regulatory region of CaMKII, a conformational change occurs in CaMKII, leading to its activation [57,76]. This activated kinase autophosphorylates the Thr286 in the regulatory domain of neighboring subunits [57,76]. Furthermore, two-photon excitation fluorescence imaging has demonstrated that glutamate stimulation recruits CaMKII into stimulated spines [77,78], induces the activation of small GTPases and actin polymerization, and eventually leads to sLTP and LTP [60,61,79,80]. The importance of CaMKII function in synaptic plasticity has been extensively studied using loss-of-function assays with inhibitory drugs, peptides, or gene silencing [56–59,81]. However, because of the lack of suitable tools, the direct effect of CaMKII activation in single spines has remained elusive. We, therefore, designed and developed paCaMKII with the idea that light could induce synaptic plasticity (i.e., sLTP) in spines.
Development of a Photoactivatable CaMKII
We aimed to develop photoactivatable CaMKII (paCaMKII) as a new optogenetic tool, which can be used to induce LTP/sLTP in a single spine by two-photon excitation. To achieve this, we opted to fuse LOV2-Jα and CaMKII genetically (Fig. 3). The light-absorbing cofactor of LOV2, FMN, has a relatively large 2-photon absorption cross-section (0.5–0.9 GM in 800–900 nm) [8], which is larger than that of other light-absorbing proteins such as Cry2 (0.02–0.04 GM in 800–900 nm, FAD) [7]. Given the availability of crystal structures of human CaMKII (PDB ID: 3SOA) [150] and LOV2-Jα (PDB ID: 2V1B) [151], we utilized them as references for our design. We first determined the insertion site for LOV2 by constructing and testing dozens of DNAs. We found that the CaMKII hinge region (residues 275–278) was suitable (Fig. 3A), i.e., in the dark state, CaMKII activity was inhibited by fusing LOV2 at this position. Next, we introduced various mutations in the regulatory domain, ensuring that the kinase and regulatory domain bind in the dark and release in the light (Figs. 3B, 3C). As a result, we successfully developed a paCaMKII. Biochemical assays confirmed that the Thr286 site of paCaMKII is autophosphorylated by light irradiation (an indication of CaMKII activity), and is dephosphorylated within a few minutes upon returning to the dark state. We also confirmed that paCaMKII is incorporated into the endogenous CaMKII dodecamer in neurons (Figs. 3D, 3E). This is not surprising since our paCaMKII has an association domain similar to that of endogenous CaMKII.

PaCaMKII Activation by Two-photon Excitation Induces sLTP and Functional LTP
To assess whether paCaMKII activation can induce sLTP within individual dendritic spines, we quantified the change in spine volume following 2-photon paCaMKII activation in single spines (Fig. 4). Biolistic gene transfer was employed to introduce a DNA plasmid encoding paCaMKII and the tdTomato (tdTomato-P2A-paCaMKII) to CA1 pyramidal neurons in cultured hippocampal slices. We assessed spine volume change by monitoring tdTomato red fluorescence by 2-photon excitation at 1000 nm. To elicit paCaMKII activation within an individual dendritic spine, we administered a low-frequency train of 2-photon excitation pulses to the spine (900 nm, 30 pulses, 1 Hz, 80 ms duration per pulse, 4 mW). Since the region where two-photon excitation occurs (~1.5 μm) and the size of the spine (<1 μm) are nearly identical, only paCaMKII in a single spine can be activated.
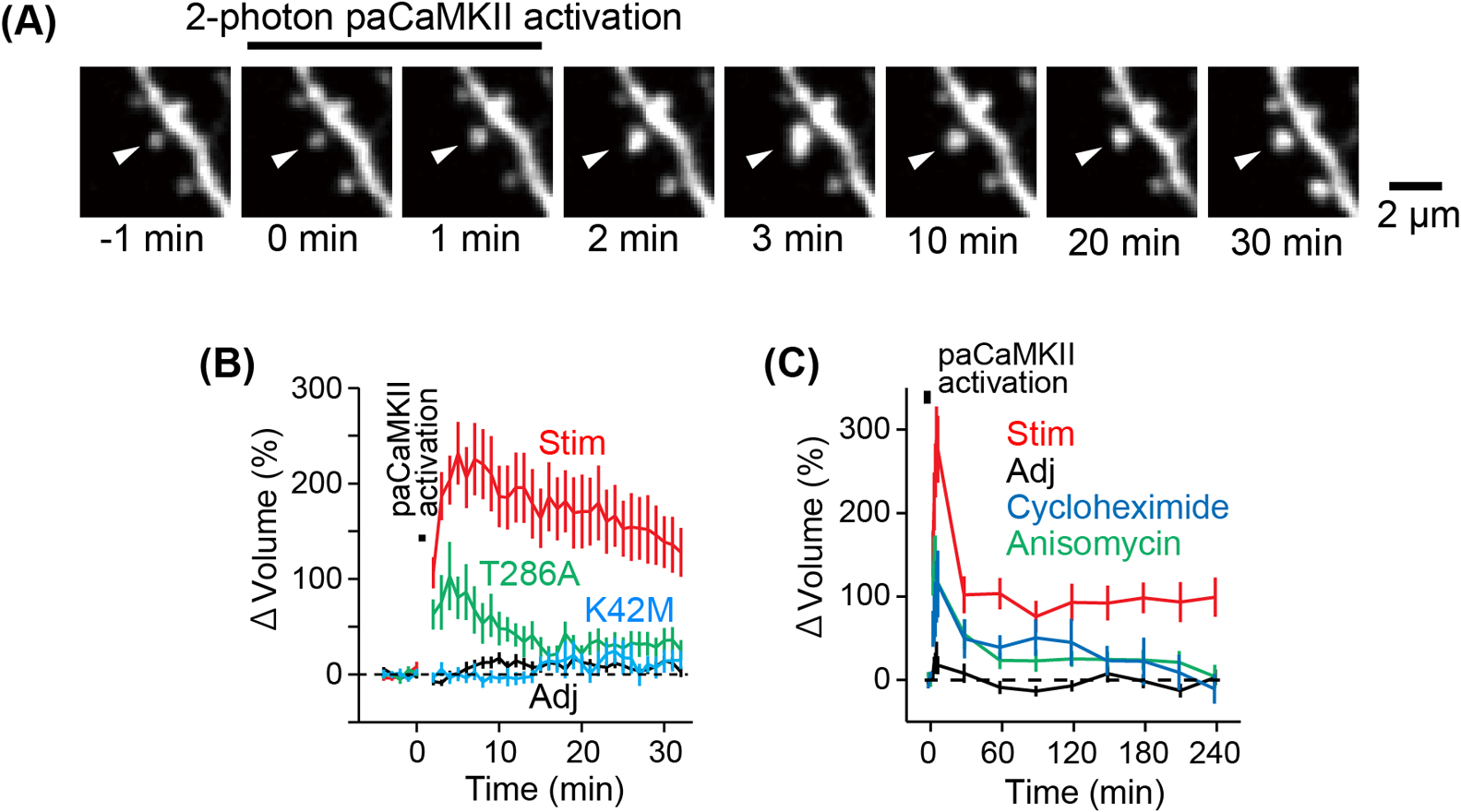
Activation of paCaMKII led to a rapid increase in spine volume (Figs. 4A, 4B), which subsequently relaxed to an elevated level of 150% after 20–30 minutes. In contrast, adjacent spines that were not stimulated by light did not exhibit an increase in volume, indicating that changes in spine volume are specific to a single spine (Fig. 4B). Long-term observation of dendritic spines after paCaMKII activation revealed that paCaMKII-induced sLTP persists for over 4 hrs (Fig. 4C). Activation of the kinase-dead mutant, K42M, did not change spine volume (Fig. 4B). Activation of the T286A autophosphorylation-deficient mutant produced a smaller and transient increase in spine volume compared to paCaMKII activation (Fig. 4B). These findings suggest that the increase in spine volume is attributable to the light-induced rise in kinase activity and autophosphorylation of paCaMKII. In addition, it is confirmed that paCaMKII activation induces spine-specific functional LTP, possibly through the recruitment of AMPAR to the stimulated spines [10].
Given that glutamate-induced sLTP may rely on protein synthesis in certain conditions [73,152], we investigated whether paCaMKII-dependent sLTP also relies on protein synthesis. Our results indicate that two different protein synthesis inhibitors, anisomycin or cycloheximide, reduced the transient volume change and inhibited persistent sLTP (Fig. 4C), suggesting that de novo protein synthesis is required for long-lasting sLTP.
LTP Induction by paCaMKII Activation in Vivo
Optogenetic tools offer significant advantages as they can be used on living animals. To verify if paCaMKII activation can trigger sLTP in vivo, we injected adeno-associated viral vectors (AAVs) encoding paCaMKII and Clover to express them in layer 2/3 neurons of the mouse cortex (Fig. 5A). For the sparse labeling of Clover, a double-floxed inverted open reading frame (DIO) system combined with low Cre expression system was used. The sparse labeling of the neurons enables us to visualize the individual spines. Using a 2-photon microscope, we imaged the neurons expressing Clover and induced sLTP at multiple spines by raster scanning in a view field with 2-photon excitation. paCaMKII activation resulted in sLTP over 30 min at multiple spines in anesthetized mice (Fig. 5B), demonstrating that paCaMKII can be used to induce sLTP in vivo.
PaCaMKII-dependent sLTP in Chronically Excited Neurons
The activity state of a neuron is well known to alter its sensitivity to synaptic inputs. For example, neurons in an aberrant neuronal excitation state increase the threshold for LTP induction [153–155]. However, it is unclear whether sLTP is also depressed in excited neurons, and molecular mechanisms of the regulation of synaptic plasticity are poorly understood.
To test if sLTP occurs in chronically excited neurons, we used two-photon paCaMKII activation [10] in cultured hippocampal slices [11]. We co-infected AAVs encoding tdTomato-P2A-paCaMKII and ESARE-d2Achilles (Fig. 6A). To label the chronically excited neurons, we utilized a synthetic activity-dependent promoter, ESARE [156], in combination with a fast-maturation mutant of yellow fluorescent proteins called Achilles [157] with a destabilization signal [158], which we referred to as d2Achilles. After 5–8 days, we incubated the slices in culture media containing bicuculline (10 μM), a GABAA receptor antagonist, for 24 hrs and successfully observed the fluorescence of d2Achilles, indicating chronically excited neurons (Figs. 6B, 6C).

To induce paCaMKII activation in single spines, we applied a low-frequency train of two-photon excitation pulses to single spines (820 nm, 30 pulses, 0.5 Hz, 80 ms duration/pulse, 4 mW). Interestingly, paCaMKII activation in chronically excited neurons failed to induce sLTP (Figs. 6D, 6E), whereas a control experiment shows spine enlargement. Since the chronic activation of neurons leads to protein synthesis [159–161], we examined whether the suppression of paCaMKII-induced sLTP requires the newly synthesized proteins during chronic neuronal activation. Notably, the inhibition of sLTP was reversed by the inhibition of protein synthesis (Figs. 6D, 6E). These results suggest that sLTP inhibition may be caused by newly synthesized proteins, inhibiting the downstream signaling of CaMKII.
Summary and Future Perspective
In this review, we have introduced recent optogenetic tools based on LOV2. These tools are highly valuable as they allow the direct activation of molecules in a complex molecular network within living cells using light. This enables us to understand their molecular functions. Many researchers have successfully developed various optogenetic tools that enable us to manipulate intracellular signaling molecules. However, most of these tools only change the localization of the active molecule by light, potentially disturbing the intracellular signalings.
Conversely, photo-activatable proteins that change their activity through light are useful since their expression rarely alters the cellular state, but not many have been developed to date. Developing such molecules requires inactivating the active site of the target molecule with LOV2 and then releasing it with light, which is challenging. Although designing molecules by referring to crystal structures is an effective method, it is almost impossible to design a molecule without one. Furthermore, even with a crystal structure, designing a molecule that suppresses molecular activity in the dark is difficult. Thus, future advances in the development of light-manipulated molecules will require breakthroughs. One strategy could be to develop and utilize protein design tools such as AlphaFold2 and other computational methods.
The most common way to photo-stimulate optogenetic tools has been through widefield illumination in combination with channelrhodopsin. However, the complexity of brain architecture has made it necessary to switch to two-photon excitation, which provides better spatial specificity and deeper penetration in scattering tissue [4,162]. For instance, single-cell resolution optogenetics based on 2-photon holographic light-targeting approaches allows the manipulation of individual cells selected by users and the generation of precise spatiotemporal neuronal activity patterns. Nevertheless, optogenetic manipulation in smaller compartments such as synapses, referred to as “Local Optogenetics”, remains challenging.
Recently, several genetically-encoded photoactivatable signaling proteins have been developed and used to study the functions of synapses [10,50,84,87,163–167]. These optogenetic tools are revolutionizing neuroscience and, more broadly, molecular cell signaling studies. In particular, optical induction of shrinkage of subsets of activated spines in vivo has been achieved recently [163,167]. Here, we introduced a photoactivatable CaMKII, which can be used to induce sLTP/LTP. As paCaMKII allows LTP induction at the level of single synapses, it should be possible to combine paCaMKII with activated-synapse tagging technology to boost the activity of specific subsets of spines in vivo. Such technology will allow the identification of the direct link between synaptic plasticity and animal behavior.
Conflict of Interest
The author declares no conflict of interest.
Author Contributions
Y.N., H.U., H.K., and H.M. wrote the manuscript.
Data Availability
The data shown in the manuscript are available from the corresponding author on reasonable request.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research in Innovative Areas (21H05703/22H02724/22H05549/23H04244/JP22H04926 Advanced Bioimaging Support (ABiS) to H.M.) from MEXT/Japan Society for the Promotion of Sciences (JSPS), Frontier Photonic Sciences Project of National Institutes of Natural Sciences (NINS) (H.M.). The Naito Foundation and the Daiko Foundation (H.M.).
References
- [1] Deisseroth, K. Optogenetics. Nat. Methods 8, 26–29 (2011). https://doi.org/10.1038/nmeth.f.324
- [2] Tischer, D., Weiner, O. D. Illuminating cell signalling with optogenetic tools. Nat. Rev. Mol. Cell Biol. 15, 551–558 (2014). https://doi.org/10.1038/nrm3837
- [3] Tye, K. M., Deisseroth, K. Optogenetic investigation of neural circuits underlying brain disease in animal models. Nat. Rev. Neurosci. 13, 251–266 (2012). https://doi.org/10.1038/nrn3171
- [4] Papagiakoumou, E., Anselmi, F., Begue, A., de Sars, V., Gluckstad, J., Isacoff, E. Y., et al. Scanless two-photon excitation of channelrhodopsin-2. Nat Methods 7, 848–854 (2010). https://doi.org/10.1038/nmeth.1505
- [5] Prakash, R., Yizhar, O., Grewe, B., Ramakrishnan, C., Wang, N., Goshen, I., et al. Two-photon optogenetic toolbox for fast inhibition, excitation and bistable modulation. Nat. Methods 9, 1171–1179 (2012). https://doi.org/10.1038/nmeth.2215
- [6] Packer, A. M., Peterka, D. S., Hirtz, J. J., Prakash, R., Deisseroth, K., Yuste, R. Two-photon optogenetics of dendritic spines and neural circuits. Nat. Methods 9, 1202–1205 (2012). https://doi.org/10.1038/nmeth.2249
- [7] Huang, S., Heikal, A. A., Webb, W. W. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys. J. 82, 2811–2825 (2002). https://doi.org/10.1016/S0006-3495(02)75621-X
- [8] Homans, R. J., Khan, R. U., Andrews, M. B., Kjeldsen, A. E., Natrajan, L. S., Marsden, S., et al. Two photon spectroscopy and microscopy of the fluorescent flavoprotein, iLOV. Phys. Chem. Chem. Phys. 20, 16949–16955 (2018). https://doi.org/10.1039/c8cp01699b
- [9] Kinjo, T., Terai, K., Horita, S., Nomura, N., Sumiyama, K., Togashi, K., et al. FRET-assisted photoactivation of flavoproteins for in vivo two-photon optogenetics. Nat. Methods 16, 1029–1036 (2019). https://doi.org/10.1038/s41592-019-0541-5
- [10] Shibata, A. C. E., Ueda, H. H., Eto, K., Onda, M., Sato, A., Ohba, T., et al. Photoactivatable CaMKII induces synaptic plasticity in single synapses. Nat. Commun. 12, 751 (2021). https://doi.org/10.1038/s41467-021-21025-6
- [11] Ueda, H. H., Nagasawa, Y., Sato, A., Onda, M., Murakoshi, H. Chronic neuronal excitation leads to dual metaplasticity in the signaling for structural long-term potentiation. Cell Rep. 38, 110153 (2022). https://doi.org/10.1016/j.celrep.2021.110153
- [12] Ueda, H. H., Nagasawa, Y., Murakoshi, H. Development of a photoactivatable CaMKII for the optical manipulation in single-synapses—local optogenetics—. SEIBUTSU BUTSURI 61, 374–377 (2021). https://doi.org/10.2142/biophys.61.374
- [13] Pudasaini, A., El-Arab, K. K., Zoltowski, B. D. LOV-based optogenetic devices: Light-driven modules to impart photoregulated control of cellular signaling. Front. Mol. Biosci. 2, 18 (2015). https://doi.org/10.3389/fmolb.2015.00018
- [14] Harper, S. M., Neil, L. C., Gardner, K. H. Structural basis of a phototropin light switch. Science 301, 1541–1544 (2003). https://doi.org/10.1126/science.1086810
- [15] Zimmerman, S. P., Kuhlman, B., Yumerefendi, H. Engineering and application of LOV2-based photoswitches. Methods Enzymol. 580, 169–190 (2016). https://doi.org/10.1016/bs.mie.2016.05.058
- [16] Lu, X., Shen, Y., Campbell, R. E. Engineering photosensory modules of non-opsin-based optogenetic actuators. Int. J. Mol. Sci. 21, 6522 (2020). https://doi.org/10.3390/ijms21186522
- [17] Wu, Y. I., Frey, D., Lungu, O. I., Jaehrig, A., Schlichting, I., Kuhlman, B., et al. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature 461, 104–108 (2009). https://doi.org/10.1038/nature08241
- [18] He, L., Tan, P., Zhu, L., Huang, K., Nguyen, N. T., Wang, R., et al. Circularly permuted LOV2 as a modular photoswitch for optogenetic engineering. Nat. Chem. Biol. 17, 915–923 (2021). https://doi.org/10.1038/s41589-021-00792-9
- [19] Geng, L., Shen, J., Wang, W. Circularly permuted AsLOV2 as an optogenetic module for engineering photoswitchable peptides. Chem. Commun. (Camb) 57, 8051–8054 (2021). https://doi.org/10.1039/d1cc02643g
- [20] Dagliyan, O., Tarnawski, M., Chu, P. H., Shirvanyants, D., Schlichting, I., Dokholyan, N. V., et al. Engineering extrinsic disorder to control protein activity in living cells. Science 354, 1441–1444 (2016). https://doi.org/10.1126/science.aah3404
- [21] Cosentino, C., Alberio, L., Gazzarrini, S., Aquila, M., Romano, E., Cermenati, S., et al. Optogenetics. Engineering of a light-gated potassium channel. Science 348, 707–710 (2015). https://doi.org/10.1126/science.aaa2787
- [22] He, L., Wang, L., Zeng, H., Tan, P., Ma, G., Zheng, S., et al. Engineering of a bona fide light-operated calcium channel. Nat. Commun. 12, 164 (2021). https://doi.org/10.1038/s41467-020-20425-4
- [23] Niopek, D., Benzinger, D., Roensch, J., Draebing, T., Wehler, P., Eils, R., et al. Engineering light-inducible nuclear localization signals for precise spatiotemporal control of protein dynamics in living cells. Nat. Commun. 5, 4404 (2014). https://doi.org/10.1038/ncomms5404
- [24] Lerner, A. M., Yumerefendi, H., Goudy, O. J., Strahl, B. D., Kuhlman, B. Engineering improved photoswitches for the control of nucleocytoplasmic distribution. ACS Synth. Biol. 7, 2898–2907 (2018). https://doi.org/10.1021/acssynbio.8b00368
- [25] Kogler, A. C., Kherdjemil, Y., Bender, K., Rabinowitz, A., Marco-Ferreres, R., Furlong, E. E. M. Extremely rapid and reversible optogenetic perturbation of nuclear proteins in living embryos. Dev. Cell 56, 2348–2363 (2021). https://doi.org/10.1016/j.devcel.2021.07.011
- [26] Strickland, D., Lin, Y., Wagner, E., Hope, C. M., Zayner, J., Antoniou, C., et al. TULIPs: Tunable, light-controlled interacting protein tags for cell biology. Nat. Methods 9, 379–384 (2012). https://doi.org/10.1038/nmeth.1904
- [27] Guntas, G., Hallett, R. A., Zimmerman, S. P., Williams, T., Yumerefendi, H., Bear, J. E., et al. Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc. Natl. Acad. Sci. U.S.A. 112, 112–117 (2015). https://doi.org/10.1073/pnas.1417910112
- [28] Wang, H., Vilela, M., Winkler, A., Tarnawski, M., Schlichting, I., Yumerefendi, H., et al. LOVTRAP: An optogenetic system for photoinduced protein dissociation. Nat. Methods 13, 755–758 (2016). https://doi.org/10.1038/nmeth.3926
- [29] Murakoshi, H., Shibata, A. C. E. ShadowY: A dark yellow fluorescent protein for FLIM-based FRET measurement. Sci. Rep. 7, 6791 (2017). https://doi.org/10.1038/s41598-017-07002-4
- [30] Zayner, J. P., Mathes, T., Sosnick, T. R., Kennis, J. T. M. Helical contributions mediate light-activated conformational change in the LOV2 domain of Avena sativa Phototropin 1. ACS Omega 4, 1238–1243 (2019). https://doi.org/10.1021/acsomega.8b02872
- [31] Renicke, C., Schuster, D., Usherenko, S., Essen, L. O., Taxis, C. A LOV2 domain-based optogenetic tool to control protein degradation and cellular function. Chem. Biol. 20, 619–626 (2013). https://doi.org/10.1016/j.chembiol.2013.03.005
- [32] Glantz, S. T., Berlew, E. E., Jaber, Z., Schuster, B. S., Gardner, K. H., Chow, B. Y. Directly light-regulated binding of RGS-LOV photoreceptors to anionic membrane phospholipids. Proc. Natl. Acad. Sci. U.S.A. 115, E7720–E7727 (2018). https://doi.org/10.1073/pnas.1802832115
- [33] Hannanta-Anan, P., Glantz, S. T., Chow, B. Y. Optically inducible membrane recruitment and signaling systems. Curr. Opin. Struct. Biol. 57, 84–92 (2019). https://doi.org/10.1016/j.sbi.2019.01.017
- [34] Berlew, E. E., Kuznetsov, I. A., Yamada, K., Bugaj, L. J., Boerckel, J. D., Chow, B. Y. Single-component optogenetic tools for inducible RhoA GTPase Signaling. Adv. Biol. (Weinh) 5, e2100810 (2021). https://doi.org/10.1002/adbi.202100810
- [35] Motta-Mena, L. B., Reade, A., Mallory, M. J., Glantz, S., Weiner, O. D., Lynch, K. W., et al. An optogenetic gene expression system with rapid activation and deactivation kinetics. Nat. Chem. Biol. 10, 196–202 (2014). https://doi.org/10.1038/nchembio.1430
- [36] Yazawa, M., Sadaghiani, A. M., Hsueh, B., Dolmetsch, R. E. Induction of protein-protein interactions in live cells using light. Nat. Biotechnol. 27, 941–945 (2009). https://doi.org/10.1038/nbt.1569
- [37] Grusch, M., Schelch, K., Riedler, R., Reichhart, E., Differ, C., Berger, W., et al. Spatio-temporally precise activation of engineered receptor tyrosine kinases by light. EMBO J. 33, 1713–1726 (2014). https://doi.org/10.15252/embj.201387695
- [38] Conrad, K. S., Bilwes, A. M., Crane, B. R. Light-induced subunit dissociation by a light-oxygen-voltage domain photoreceptor from Rhodobacter sphaeroides. Biochemistry 52, 378–391 (2013). https://doi.org/10.1021/bi3015373
- [39] Richter, F., Fonfara, I., Bouazza, B., Schumacher, C. H., Bratovic, M., Charpentier, E., et al. Engineering of temperature- and light-switchable Cas9 variants. Nucleic Acids Res. 44, gkw930 (2016). https://doi.org/10.1093/nar/gkw930
- [40] Moglich, A., Moffat, K. Structural basis for light-dependent signaling in the dimeric LOV domain of the photosensor YtvA. J. Mol. Biol. 373, 112–126 (2007). https://doi.org/10.1016/j.jmb.2007.07.039
- [41] Moglich, A., Ayers, R. A., Moffat, K. Design and signaling mechanism of light-regulated histidine kinases. J. Mol. Biol. 385, 1433–1444 (2009). https://doi.org/10.1016/j.jmb.2008.12.017
- [42] Wang, X., Chen, X., Yang, Y. Spatiotemporal control of gene expression by a light-switchable transgene system. Nat. Methods 9, 266–269 (2012). https://doi.org/10.1038/nmeth.1892
- [43] Kawano, F., Suzuki, H., Furuya, A., Sato, M. Engineered pairs of distinct photoswitches for optogenetic control of cellular proteins. Nat. Commun. 6, 6256 (2015). https://doi.org/10.1038/ncomms7256
- [44] Salinas, F., Rojas, V., Delgado, V., Lopez, J., Agosin, E., Larrondo, L. F. Fungal light-oxygen-voltage domains for optogenetic control of gene expression and flocculation in yeast. mBio 9, e00626-18 (2018). https://doi.org/10.1128/mBio.00626-18
- [45] Che, D. L., Duan, L., Zhang, K., Cui, B. The dual characteristics of light-induced Cryptochrome 2, homo-oligomerization and heterodimerization, for optogenetic manipulation in mammalian cells. ACS Synth. Biol. 4, 1124–1135 (2015). https://doi.org/10.1021/acssynbio.5b00048
- [46] Duan, L., Hope, J., Ong, Q., Lou, H. Y., Kim, N., McCarthy, C., et al. Understanding CRY2 interactions for optical control of intracellular signaling. Nat. Commun. 8, 547 (2017). https://doi.org/10.1038/s41467-017-00648-8
- [47] Liu, H., Yu, X., Li, K., Klejnot, J., Yang, H., Lisiero, D., et al. Photoexcited CRY2 interacts with CIB1 to regulate transcription and floral initiation in Arabidopsis. Science 322, 1535–1539 (2008). https://doi.org/10.1126/science.1163927
- [48] Kennedy, M. J., Hughes, R. M., Peteya, L. A., Schwartz, J. W., Ehlers, M. D., Tucker, C. L. Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods 7, 973–975 (2010). https://doi.org/10.1038/nmeth.1524
- [49] Taslimi, A., Zoltowski, B., Miranda, J. G., Pathak, G. P., Hughes, R. M., Tucker, C. L. Optimized second-generation CRY2-CIB dimerizers and photoactivatable Cre recombinase. Nat. Chem. Biol. 12, 425–430 (2016). https://doi.org/10.1038/nchembio.2063
- [50] Sinnen, B. L., Bowen, A. B., Forte, J. S., Hiester, B. G., Crosby, K. C., Gibson, E. S., et al. Optogenetic control of synaptic composition and function. Neuron 93, 646–660 (2017). https://doi.org/10.1016/j.neuron.2016.12.037
- [51] Bugaj, L. J., Choksi, A. T., Mesuda, C. K., Kane, R. S., Schaffer, D. V. Optogenetic protein clustering and signaling activation in mammalian cells. Nat. Methods 10, 249–252 (2013). https://doi.org/10.1038/nmeth.2360
- [52] Taslimi, A., Vrana, J. D., Chen, D., Borinskaya, S., Mayer, B. J., Kennedy, M. J., et al. An optimized optogenetic clustering tool for probing protein interaction and function. Nat. Commun. 5, 4925 (2014). https://doi.org/10.1038/ncomms5925
- [53] Park, H., Kim, N. Y., Lee, S., Kim, N., Kim, J., Heo, W. D. Optogenetic protein clustering through fluorescent protein tagging and extension of CRY2. Nat. Commun. 8, 30 (2017). https://doi.org/10.1038/s41467-017-00060-2
- [54] Nicoll, R. A. A brief history of long-term potentiation. Neuron 93, 281–290 (2017). https://doi.org/10.1016/j.neuron.2016.12.015
- [55] Yashiro, K., Philpot, B. D. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 55, 1081–1094 (2008). https://doi.org/10.1016/j.neuropharm.2008.07.046
- [56] Lisman, J., Yasuda, R., Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 13, 169–182 (2012). https://doi.org/10.1038/nrn3192
- [57] Bayer, K. U., Schulman, H. CaM kinase: Still inspiring at 40. Neuron 103, 380–394 (2019). https://doi.org/10.1016/j.neuron.2019.05.033
- [58] Giese, K. P., Mizuno, K. The roles of protein kinases in learning and memory. Learn. Mem. 20, 540–552 (2013). https://doi.org/10.1101/lm.028449.112
- [59] Herring, B. E., Nicoll, R. A. Long-term potentiation: From CaMKII to AMPA receptor trafficking. Annu. Rev. Physiol. 78, 351–365 (2016). https://doi.org/10.1146/annurev-physiol-021014-071753
- [60] Lee, S. J., Escobedo-Lozoya, Y., Szatmari, E. M., Yasuda, R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 458, 299–304 (2009). https://doi.org/10.1038/nature07842
- [61] Saneyoshi, T., Matsuno, H., Suzuki, A., Murakoshi, H., Hedrick, N. G., Agnello, E., et al. Reciprocal activation within a kinase-effector complex underlying persistence of structural LTP. Neuron 102, 1199–1210 (2019). https://doi.org/10.1016/j.neuron.2019.04.012
- [62] Murakoshi, H., Yasuda, R. Postsynaptic signaling during plasticity of dendritic spines. Trends Neurosci. 35, 135–143 (2012). https://doi.org/10.1016/j.tins.2011.12.002
- [63] Bosch, M., Castro, J., Saneyoshi, T., Matsuno, H., Sur, M., Hayashi, Y. Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 82, 444–459 (2014). https://doi.org/10.1016/j.neuron.2014.03.021
- [64] Nakahata, Y., Yasuda, R. Plasticity of spine structure: Local signaling, translation and cytoskeletal reorganization. Front. Synaptic. Neurosci. 10, 29 (2018). https://doi.org/10.3389/fnsyn.2018.00029
- [65] Malinow, R., Malenka, R. C. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126 (2002). https://doi.org/10.1146/annurev.neuro.25.112701.142758
- [66] Derkach, V. A., Oh, M. C., Guire, E. S., Soderling, T. R. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat. Rev. Neurosci. 8, 101–113 (2007). https://doi.org/10.1038/nrn2055
- [67] Cingolani, L. A., Goda, Y. Actin in action: The interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci. 9, 344–356 (2008). https://doi.org/10.1038/nrn2373
- [68] Pettit, D. L., Perlman, S., Malinow, R. Potentiated transmission and prevention of further LTP by increased CaMKII activity in postsynaptic hippocampal slice neurons. Science 266, 1881–1885 (1994). https://doi.org/10.1126/science.7997883
- [69] Lledo, P. M., Hjelmstad, G. O., Mukherji, S., Soderling, T. R., Malenka, R. C., Nicoll, R. A. Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc. Natl. Acad. Sci. U.S.A. 92, 11175–11179 (1995). https://doi.org/10.1073/pnas.92.24.11175
- [70] Jourdain, P., Fukunaga, K., Muller, D. Calcium/calmodulin-dependent protein kinase II contributes to activity-dependent filopodia growth and spine formation. J. Neurosci. 23, 10645–10649 (2003). https://doi.org/10.1523/JNEUROSCI.23-33-10645.2003
- [71] Matsuzaki, M., Honkura, N., Ellis-Davies, G. C., Kasai, H. Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766 (2004). https://doi.org/10.1038/nature02617
- [72] Harvey, C. D., Svoboda, K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature 450, 1195–1200 (2007). https://doi.org/10.1038/nature06416
- [73] Govindarajan, A., Israely, I., Huang, S. Y., Tonegawa, S. The dendritic branch is the preferred integrative unit for protein synthesis-dependent LTP. Neuron 69, 132–146 (2011). https://doi.org/10.1016/j.neuron.2010.12.008
- [74] McGuinness, T. L., Lai, Y., Greengard, P. Ca2+/calmodulin-dependent protein kinase II. Isozymic forms from rat forebrain and cerebellum. J. Biol. Chem. 260, 1696–1704 (1985).
- [75] Myers, J. B., Zaegel, V., Coultrap, S. J., Miller, A. P., Bayer, K. U., Reichow, S. L. The CaMKII holoenzyme structure in activation-competent conformations. Nat. Commun. 8, 15742 (2017). https://doi.org/10.1038/ncomms15742
- [76] Lisman, J., Schulman, H., Cline, H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 3, 175–190 (2002). https://doi.org/10.1038/nrn753
- [77] Bayer, K. U., LeBel, E., McDonald, G. L., O’Leary, H., Schulman, H., De Koninck, P. Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J. Neurosci. 26, 1164–1174 (2006). https://doi.org/10.1523/JNEUROSCI.3116-05.2006
- [78] Zhang, Y. P., Holbro, N., Oertner, T. G. Optical induction of plasticity at single synapses reveals input-specific accumulation of αCaMKII. Proc. Natl. Acad. Sci. U.S.A. 105, 12039–12044 (2008). https://doi.org/10.1073/pnas.0802940105
- [79] Murakoshi, H., Wang, H., Yasuda, R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 472, 100–104 (2011). https://doi.org/10.1038/nature09823
- [80] Hedrick, N. G., Harward, S. C., Hall, C. E., Murakoshi, H., McNamara, J. O., Yasuda, R. Rho GTPase complementation underlies BDNF-dependent homo- and heterosynaptic plasticity. Nature 538, 104–108 (2016). https://doi.org/10.1038/nature19784
- [81] Coultrap, S. J., Bayer, K. U. CaMKII regulation in information processing and storage. Trends Neurosci. 35, 607–618 (2012). https://doi.org/10.1016/j.tins.2012.05.003
- [82] Carrasco-Lopez, C., Zhao, E. M., Gil, A. A., Alam, N., Toettcher, J. E., Avalos, J. L. Development of light-responsive protein binding in the monobody non-immunoglobulin scaffold. Nat. Commun. 11, 4045 (2020). https://doi.org/10.1038/s41467-020-17837-7
- [83] Gil, A. A., Carrasco-Lopez, C., Zhu, L., Zhao, E. M., Ravindran, P. T., Wilson, M. Z., et al. Optogenetic control of protein binding using light-switchable nanobodies. Nat. Commun. 11, 4044 (2020). https://doi.org/10.1038/s41467-020-17836-8
- [84] Murakoshi, H., Shin, M. E., Parra-Bueno, P., Szatmari, E. M., Shibata, A. C. E., Yasuda, R. Kinetics of endogenous CaMKII required for synaptic plasticity revealed by optogenetic kinase inhibitor. Neuron 94, 37–47 (2017). https://doi.org/10.1016/j.neuron.2017.02.036
- [85] Yi, J. J., Wang, H., Vilela, M., Danuser, G., Hahn, K. M. Manipulation of endogenous kinase activity in living cells using photoswitchable inhibitory peptides. ACS Synth. Biol. 3, 788–795 (2014). https://doi.org/10.1021/sb5001356
- [86] Melero-Fernandez de Mera, R. M., Li, L. L., Popinigis, A., Cisek, K., Tuittila, M., Yadav, L., et al. A simple optogenetic MAPK inhibitor design reveals resonance between transcription-regulating circuitry and temporally-encoded inputs. Nat. Commun. 8, 15017 (2017). https://doi.org/10.1038/ncomms15017
- [87] Hollos, P., John, J. M., Lehtonen, J. V., Coffey, E. T. Optogenetic control of spine-head JNK reveals a role in dendritic spine regression. eNeuro 7, 0303–0319 (2020). https://doi.org/10.1523/ENEURO.0303-19.2019
- [88] Bonger, K. M., Rakhit, R., Payumo, A. Y., Chen, J. K., Wandless, T. J. General method for regulating protein stability with light. ACS Chem. Biol. 9, 111–115 (2014). https://doi.org/10.1021/cb400755b
- [89] Hasenjager, S., Trauth, J., Hepp, S., Goenrich, J., Essen, L. O., Taxis, C. Optogenetic downregulation of protein levels with an ultrasensitive switch. ACS Synth. Biol. 8, 1026–1036 (2019). https://doi.org/10.1021/acssynbio.8b00471
- [90] Spiltoir, J. I., Strickland, D., Glotzer, M., Tucker, C. L. Optical control of peroxisomal trafficking. ACS Synth. Biol. 5, 554–560 (2016). https://doi.org/10.1021/acssynbio.5b00144
- [91] Nakamura, M., Chen, L., Howes, S. C., Schindler, T. D., Nogales, E., Bryant, Z. Remote control of myosin and kinesin motors using light-activated gearshifting. Nat. Nanotechnol. 9, 693–697 (2014). https://doi.org/10.1038/nnano.2014.147
- [92] Wang, W., Wildes, C. P., Pattarabanjird, T., Sanchez, M. I., Glober, G. F., Matthews, G. A., et al. A light- and calcium-gated transcription factor for imaging and manipulating activated neurons. Nat. Biotechnol. 35, 864–871 (2017). https://doi.org/10.1038/nbt.3909
- [93] Lee, D., Hyun, J. H., Jung, K., Hannan, P., Kwon, H. B. A calcium- and light-gated switch to induce gene expression in activated neurons. Nat. Biotechnol. 35, 858–863 (2017). https://doi.org/10.1038/nbt.3902
- [94] Kim, M. W., Wang, W., Sanchez, M. I., Coukos, R., von Zastrow, M., Ting, A. Y. Time-gated detection of protein-protein interactions with transcriptional readout. eLife 6, 30233 (2017). https://doi.org/10.7554/eLife.30233
- [95] Sanchez, M. I., Ting, A. Y. Directed evolution improves the catalytic efficiency of TEV protease. Nat. Methods 17, 167–174 (2020). https://doi.org/10.1038/s41592-019-0665-7
- [96] Duplus-Bottin, H., Spichty, M., Triqueneaux, G., Place, C., Mangeot, P. E., Ohlmann, T., et al. A single-chain and fast-responding light-inducible Cre recombinase as a novel optogenetic switch. eLife 10, 61268 (2021). https://doi.org/10.7554/eLife.61268
- [97] Bubeck, F., Hoffmann, M. D., Harteveld, Z., Aschenbrenner, S., Bietz, A., Waldhauer, M. C., et al. Engineered anti-CRISPR proteins for optogenetic control of CRISPR-Cas9. Nat. Methods 15, 924–927 (2018). https://doi.org/10.1038/s41592-018-0178-9
- [98] Hoffmann, M. D., Mathony, J., Upmeier Zu Belzen, J., Harteveld, Z., Aschenbrenner, S., Stengl, C., et al. Optogenetic control of Neisseria meningitidis Cas9 genome editing using an engineered, light-switchable anti-CRISPR protein. Nucleic Acids. Res. 49, e29 (2021). https://doi.org/10.1093/nar/gkaa1198
- [99] Schierling, B., Pingoud, A. Controlling the DNA cleavage activity of light-inducible chimeric endonucleases by bidirectional photoactivation. Bioconjug. Chem. 23, 1105–1109 (2012). https://doi.org/10.1021/bc3001326
- [100] Seifert, S., Ehrt, C., Luckfeldt, L., Lubeck, M., Schramm, F., Brakmann, S. Optical control of transcription: Genetically encoded photoswitchable variants of T7 RNA polymerase. Chembiochem 20, 2813–2817 (2019). https://doi.org/10.1002/cbic.201900298
- [101] Smart, A. D., Pache, R. A., Thomsen, N. D., Kortemme, T., Davis, G. W., Wells, J. A. Engineering a light-activated caspase-3 for precise ablation of neurons in vivo. Proc. Natl. Acad. Sci. U.S.A. 114, E8174–E8183 (2017). https://doi.org/10.1073/pnas.1705064114
- [102] Li, L., He, L., Wu, B., Yu, C., Zhao, H., Zhou, Y., et al. Structural determinants for light-dependent membrane binding of a photoswitchable polybasic domain. ACS Synth. Biol. 10, 542–551 (2021). https://doi.org/10.1021/acssynbio.0c00571
- [103] van Bergeijk, P., Adrian, M., Hoogenraad, C. C., Kapitein, L. C. Optogenetic control of organelle transport and positioning. Nature 518, 111–114 (2015). https://doi.org/10.1038/nature14128
- [104] Oakes, P. W., Wagner, E., Brand, C. A., Probst, D., Linke, M., Schwarz, U. S., et al. Optogenetic control of RhoA reveals zyxin-mediated elasticity of stress fibres. Nat. Commun. 8, 15817 (2017). https://doi.org/10.1038/ncomms15817
- [105] Cavanaugh, K. E., Staddon, M. F., Munro, E., Banerjee, S., Gardel, M. L. RhoA mediates epithelial cell shape changes via mechanosensitive endocytosis. Dev. Cell 52, 152–166 (2020). https://doi.org/10.1016/j.devcel.2019.12.002
- [106] Johnson, H. E., Goyal, Y., Pannucci, N. L., Schupbach, T., Shvartsman, S. Y., Toettcher, J. E. The spatiotemporal limits of developmental Erk signaling. Dev. Cell 40, 185–192 (2017). https://doi.org/10.1016/j.devcel.2016.12.002
- [107] Inaba, H., Miao, Q., Nakata, T. Optogenetic control of small GTPases reveals RhoA mediates intracellular calcium signaling. J. Biol. Chem. 296, 100290 (2021). https://doi.org/10.1016/j.jbc.2021.100290
- [108] Rich, A., Fehon, R. G., Glotzer, M. Rho1 activation recapitulates early gastrulation events in the ventral, but not dorsal, epithelium of Drosophila embryos. eLife 9, 56893 (2020). https://doi.org/10.7554/eLife.56893
- [109] Adikes, R. C., Hallett, R. A., Saway, B. F., Kuhlman, B., Slep, K. C. Control of microtubule dynamics using an optogenetic microtubule plus end-F-actin cross-linker. J. Cell Biol. 217, 779–793 (2018). https://doi.org/10.1083/jcb.201705190
- [110] Ma, G., Liu, J., Ke, Y., Liu, X., Li, M., Wang, F., et al. Optogenetic control of voltage-gated calcium channels. Angew. Chem. Int. Ed. Engl. 57, 7019–7022 (2018). https://doi.org/10.1002/anie.201713080
- [111] Shi, F., Kawano, F., Park, S. E., Komazaki, S., Hirabayashi, Y., Polleux, F., et al. Optogenetic control of endoplasmic reticulum-mitochondria tethering. ACS Synth. Biol. 7, 2–9 (2018). https://doi.org/10.1021/acssynbio.7b00248
- [112] Bracha, D., Walls, M. T., Wei, M. T., Zhu, L., Kurian, M., Avalos, J. L., et al. Mapping local and global liquid phase behavior in living cells using photo-oligomerizable seeds. Cell 175, 1467–1480 (2018). https://doi.org/10.1016/j.cell.2018.10.048
- [113] Shin, Y., Chang, Y. C., Lee, D. S. W., Berry, J., Sanders, D. W., Ronceray, P., et al. Liquid nuclear condensates mechanically sense and restructure the genome. Cell 175, 1481–1491 (2018). https://doi.org/10.1016/j.cell.2018.10.057
- [114] D’Acunzo, P., Strappazzon, F., Caruana, I., Meneghetti, G., Di Rita, A., Simula, L., et al. Reversible induction of mitophagy by an optogenetic bimodular system. Nat. Commun. 10, 1533 (2019). https://doi.org/10.1038/s41467-019-09487-1
- [115] Yamamoto, K., Miura, H., Ishida, M., Mii, Y., Kinoshita, N., Takada, S., et al. Optogenetic relaxation of actomyosin contractility uncovers mechanistic roles of cortical tension during cytokinesis. Nat. Commun. 12, 7145 (2021). https://doi.org/10.1038/s41467-021-27458-3
- [116] Nijenhuis, W., van Grinsven, M. M. P., Kapitein, L. C. An optimized toolbox for the optogenetic control of intracellular transport. J. Cell Biol. 219, e20197149 (2020). https://doi.org/10.1083/jcb.201907149
- [117] van Haren, J., Charafeddine, R. A., Ettinger, A., Wang, H., Hahn, K. M., Wittmann, T. Local control of intracellular microtubule dynamics by EB1 photodissociation. Nat. Cell Biol. 20, 252–261 (2018). https://doi.org/10.1038/s41556-017-0028-5
- [118] Takano, T., Wu, M., Nakamuta, S., Naoki, H., Ishizawa, N., Namba, T., et al. Discovery of long-range inhibitory signaling to ensure single axon formation. Nat. Commun. 8, 33 (2017). https://doi.org/10.1038/s41467-017-00044-2
- [119] Shcherbakova, D. M., Cox Cammer, N., Huisman, T. M., Verkhusha, V. V., Hodgson, L. Direct multiplex imaging and optogenetics of Rho GTPases enabled by near-infrared FRET. Nat. Chem. Biol. 14, 591–600 (2018). https://doi.org/10.1038/s41589-018-0044-1
- [120] Stone, O. J., Pankow, N., Liu, B., Sharma, V. P., Eddy, R. J., Wang, H., et al. Optogenetic control of cofilin and αTAT in living cells using Z-lock. Nat. Chem. Biol. 15, 1183–1190 (2019). https://doi.org/10.1038/s41589-019-0405-4
- [121] Benman, W., Berlew, E. E., Deng, H., Parker, C., Kuznetsov, I. A., Lim, B., et al. Temperature-responsive optogenetic probes of cell signaling. Nat. Chem. Biol. 18, 152–160 (2022). https://doi.org/10.1038/s41589-021-00917-0
- [122] Berlew, E. E., Yamada, K., Kuznetsov, I. A., Rand, E. A., Ochs, C. C., Jaber, Z., et al. Designing single-component optogenetic membrane recruitment systems: The Rho-family GTPase signaling toolbox. ACS Synth. Biol. 11, 515–521 (2022). https://doi.org/10.1021/acssynbio.1c00604
- [123] Baaske, J., Gonschorek, P., Engesser, R., Dominguez-Monedero, A., Raute, K., Fischbach, P., et al. Dual-controlled optogenetic system for the rapid down-regulation of protein levels in mammalian cells. Sci. Rep. 8, 15024 (2018). https://doi.org/10.1038/s41598-018-32929-7
- [124] Polstein, L. R., Gersbach, C. A. Light-inducible spatiotemporal control of gene activation by customizable zinc finger transcription factors. J. Am. Chem. Soc. 134, 16480–16483 (2012). https://doi.org/10.1021/ja3065667
- [125] Dixon, R. E., Yuan, C., Cheng, E. P., Navedo, M. F., Santana, L. F. Ca2+ signaling amplification by oligomerization of L-type Cav1.2 channels. Proc. Natl. Acad. Sci. U.S.A. 109, 1749–1754 (2012). https://doi.org/10.1073/pnas.1116731109
- [126] Khamo, J. S., Krishnamurthy, V. V., Chen, Q., Diao, J., Zhang, K. Optogenetic delineation of receptor tyrosine kinase subcircuits in PC12 cell differentiation. Cell Chem. Biol. 26, 400–410 (2019). https://doi.org/10.1016/j.chembiol.2018.11.004
- [127] Huang, T., Zhang, Y., Wang, Z., Zeng, Y., Wang, N., Fan, H., et al. Optogenetically controlled TrkA activity improves the regenerative capacity of hair-follicle-derived stem cells to differentiate into neurons and glia. Adv. Biol. (Weinh) 5, e2000134 (2021). https://doi.org/10.1002/adbi.202000134
- [128] Sako, K., Pradhan, S. J., Barone, V., Ingles-Prieto, A., Muller, P., Ruprecht, V., et al. Optogenetic control of Nodal signaling reveals a temporal pattern of Nodal signaling regulating cell fate specification during gastrulation. Cell Rep. 16, 866–877 (2016). https://doi.org/10.1016/j.celrep.2016.06.036
- [129] Jerng, H. H., Patel, J. M., Khan, T. A., Arenkiel, B. R., Pfaffinger, P. J. Light-regulated voltage-gated potassium channels for acute interrogation of channel function in neurons and behavior. PLoS One 16, e0248688 (2021). https://doi.org/10.1371/journal.pone.0248688
- [130] Li, X., Zhang, C., Xu, X., Miao, J., Yao, J., Liu, R., et al. A single-component light sensor system allows highly tunable and direct activation of gene expression in bacterial cells. Nucleic Acids. Res. 48, e33 (2020). https://doi.org/10.1093/nar/gkaa044
- [131] Ohlendorf, R., Vidavski, R. R., Eldar, A., Moffat, K., Moglich, A. From dusk till dawn: one-plasmid systems for light-regulated gene expression. J. Mol. Biol. 416, 534–542 (2012). https://doi.org/10.1016/j.jmb.2012.01.001
- [132] Lalwani, M. A., Ip, S. S., Carrasco-Lopez, C., Day, C., Zhao, E. M., Kawabe, H., et al. Optogenetic control of the lac operon for bacterial chemical and protein production. Nat. Chem. Biol. 17, 71–79 (2021). https://doi.org/10.1038/s41589-020-0639-1
- [133] Lalwani, M. A., Kawabe, H., Mays, R. L., Hoffman, S. M., Avalos, J. L. Optogenetic control of microbial consortia populations for chemical production. ACS Synth. Biol. 10, 2015–2029 (2021). https://doi.org/10.1021/acssynbio.1c00182
- [134] Cheng, X., Pu, L., Fu, S., Xia, A., Huang, S., Ni, L., et al. Engineering Gac/Rsm signaling cascade for optogenetic induction of the pathogenicity switch in Pseudomonas aeruginosa. ACS Synth. Biol. 10, 1520–1530 (2021). https://doi.org/10.1021/acssynbio.1c00075
- [135] Li, T., Chen, X., Qian, Y., Shao, J., Li, X., Liu, S., et al. A synthetic BRET-based optogenetic device for pulsatile transgene expression enabling glucose homeostasis in mice. Nat. Commun. 12, 615 (2021). https://doi.org/10.1038/s41467-021-20913-1
- [136] Liu, R., Yang, J., Yao, J., Zhao, Z., He, W., Su, N., et al. Optogenetic control of RNA function and metabolism using engineered light-switchable RNA-binding proteins. Nat. Biotechnol. 40, 779–786 (2022). https://doi.org/10.1038/s41587-021-01112-1
- [137] Sheets, M. B., Wong, W. W., Dunlop, M. J. Light-inducible recombinases for bacterial optogenetics. ACS Synth. Biol. 9, 227–235 (2020). https://doi.org/10.1021/acssynbio.9b00395
- [138] Yao, S., Yuan, P., Ouellette, B., Zhou, T., Mortrud, M., Balaram, P., et al. RecV recombinase system for in vivo targeted optogenomic modifications of single cells or cell populations. Nat. Methods 17, 422–429 (2020). https://doi.org/10.1038/s41592-020-0774-3
- [139] Shaaya, M., Fauser, J., Zhurikhina, A., Conage-Pough, J. E., Huyot, V., Brennan, M., et al. Light-regulated allosteric switch enables temporal and subcellular control of enzyme activity. eLife 9, 60647 (2020). https://doi.org/10.7554/eLife.60647
- [140] Aper, S. J., Merkx, M. Rewiring multidomain protein switches: Transforming a fluorescent Zn2+ sensor into a light-responsive Zn2+ binding protein. ACS Synth. Biol. 5, 698–709 (2016). https://doi.org/10.1021/acssynbio.6b00027
- [141] di Pietro, F., Herszterg, S., Huang, A., Bosveld, F., Alexandre, C., Sancere, L., et al. Rapid and robust optogenetic control of gene expression in Drosophila. Dev. Cell 56, 3393–3404 (2021). https://doi.org/10.1016/j.devcel.2021.11.016
- [142] Baumschlager, A., Aoki, S. K., Khammash, M. Dynamic blue light-inducible T7 RNA polymerases (Opto-T7RNAPs) for precise spatiotemporal gene expression control. ACS Synth. Biol. 6, 2157–2167 (2017). https://doi.org/10.1021/acssynbio.7b00169
- [143] Kawano, F., Okazaki, R., Yazawa, M., Sato, M. A photoactivatable Cre-loxP recombination system for optogenetic genome engineering. Nat. Chem. Biol. 12, 1059–1064 (2016). https://doi.org/10.1038/nchembio.2205
- [144] Morikawa, K., Furuhashi, K., de Sena-Tomas, C., Garcia-Garcia, A. L., Bekdash, R., Klein, A. D., et al. Photoactivatable Cre recombinase 3.0 for in vivo mouse applications. Nat. Commun. 11, 2141 (2020). https://doi.org/10.1038/s41467-020-16030-0
- [145] Nihongaki, Y., Kawano, F., Nakajima, T., Sato, M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 33, 755–760 (2015). https://doi.org/10.1038/nbt.3245
- [146] Nihongaki, Y., Furuhata, Y., Otabe, T., Hasegawa, S., Yoshimoto, K., Sato, M. CRISPR-Cas9-based photoactivatable transcription systems to induce neuronal differentiation. Nat. Methods 14, 963–966 (2017). https://doi.org/10.1038/nmeth.4430
- [147] Benedetti, L., Marvin, J. S., Falahati, H., Guillen-Samander, A., Looger, L. L., De Camilli, P. Optimized Vivid-derived Magnets photodimerizers for subcellular optogenetics in mammalian cells. eLife 9, 63230 (2020). https://doi.org/10.7554/eLife.63230
- [148] Chen, F., Wegner, S. V. Blue-light-switchable bacterial cell-cell adhesions enable the control of multicellular bacterial communities. ACS Synth. Biol. 9, 1169–1180 (2020). https://doi.org/10.1021/acssynbio.0c00054
- [149] Yu, D., Lee, H., Hong, J., Jung, H., Jo, Y., Oh, B. H., et al. Optogenetic activation of intracellular antibodies for direct modulation of endogenous proteins. Nat. Methods 16, 1095–1100 (2019). https://doi.org/10.1038/s41592-019-0592-7
- [150] Chao, L. H., Stratton, M. M., Lee, I. H., Rosenberg, O. S., Levitz, J., Mandell, D. J., et al. A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin-dependent kinase II holoenzyme. Cell 146, 732–745 (2011). https://doi.org/10.1016/j.cell.2011.07.038
- [151] Halavaty, A. S., Moffat, K. N- and C-terminal flanking regions modulate light-induced signal transduction in the LOV2 domain of the blue light sensor phototropin 1 from Avena sativa. Biochemistry 46, 14001–14009 (2007). https://doi.org/10.1021/bi701543e
- [152] Tanaka, J., Horiike, Y., Matsuzaki, M., Miyazaki, T., Ellis-Davies, G. C., Kasai, H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science 319, 1683–1687 (2008). https://doi.org/10.1126/science.1152864
- [153] Abraham, W. C. Metaplasticity: Tuning synapses and networks for plasticity. Nat. Rev. Neurosci. 9, 387 (2008). https://doi.org/10.1038/nrn2356
- [154] Cooper, L. N., Bear, M. F. The BCM theory of synapse modification at 30: interaction of theory with experiment. Nat. Rev. Neurosci. 13, 798–810 (2012). https://doi.org/10.1038/nrn3353
- [155] Keck, T., Hubener, M., Bonhoeffer, T. Interactions between synaptic homeostatic mechanisms: an attempt to reconcile BCM theory, synaptic scaling, and changing excitation/inhibition balance. Curr. Opin. Neurobiol. 43, 87–93 (2017). https://doi.org/10.1016/j.conb.2017.02.003
- [156] Kawashima, T., Kitamura, K., Suzuki, K., Nonaka, M., Kamijo, S., Takemoto-Kimura, S., et al. Functional labeling of neurons and their projections using the synthetic activity-dependent promoter E-SARE. Nat. Methods 10, 889–895 (2013). https://doi.org/10.1038/nmeth.2559
- [157] Yoshioka-Kobayashi, K., Matsumiya, M., Niino, Y., Isomura, A., Kori, H., Miyawaki, A., et al. Coupling delay controls synchronized oscillation in the segmentation clock. Nature 580, 119–123 (2020). https://doi.org/10.1038/s41586-019-1882-z
- [158] Li, X., Zhao, X., Fang, Y., Jiang, X., Duong, T., Fan, C., et al. Generation of destabilized green fluorescent protein as a transcription reporter. J. Biol. Chem. 273, 34970–34975 (1998). https://doi.org/10.1074/jbc.273.52.34970
- [159] Schanzenbacher, C. T., Sambandan, S., Langer, J. D., Schuman, E. M. Nascent proteome remodeling following homeostatic scaling at hippocampal synapses. Neuron 92, 358–371 (2016). https://doi.org/10.1016/j.neuron.2016.09.058
- [160] Schanzenbacher, C. T., Langer, J. D., Schuman, E. M. Time- and polarity-dependent proteomic changes associated with homeostatic scaling at central synapses. eLife 7, 33322 (2018). https://doi.org/10.7554/eLife.33322
- [161] Dorrbaum, A. R., Alvarez-Castelao, B., Nassim-Assir, B., Langer, J. D., Schuman, E. M. Proteome dynamics during homeostatic scaling in cultured neurons. eLife 9, 52939 (2020). https://doi.org/10.7554/eLife.52939
- [162] Andrasfalvy, B. K., Zemelman, B. V., Tang, J., Vaziri, A. Two-photon single-cell optogenetic control of neuronal activity by sculpted light. Proc. Natl. Acad. Sci. U.S.A. 107, 11981–11986 (2010). https://doi.org/10.1073/pnas.1006620107
- [163] Hayashi-Takagi, A., Yagishita, S., Nakamura, M., Shirai, F., Wu, Y. I., Loshbaugh, A. L., et al. Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 525, 333–338 (2015). https://doi.org/10.1038/nature15257
- [164] Locke, C., Machida, K., Tucker, C. L., Wu, Y., Yu, J. Optogenetic activation of EphB2 receptor in dendrites induced actin polymerization by activating Arg kinase. Biol. Open 6, 1820–1830 (2017). https://doi.org/10.1242/bio.029900
- [165] Kakegawa, W., Katoh, A., Narumi, S., Miura, E., Motohashi, J., Takahashi, A., et al. Optogenetic control of synaptic AMPA receptor endocytosis reveals roles of LTD in motor learning. Neuron 99, 985–998 (2018). https://doi.org/10.1016/j.neuron.2018.07.034
- [166] Letellier, M., Lagardere, M., Tessier, B., Janovjak, H., Thoumine, O. Optogenetic control of excitatory post-synaptic differentiation through neuroligin-1 tyrosine phosphorylation. eLife 9, 52027 (2020). https://doi.org/10.7554/eLife.52027
- [167] Goto, A., Bota, A., Miya, K., Wang, J., Tsukamoto, S., Jiang, X., et al. Stepwise synaptic plasticity events drive the early phase of memory consolidation. Science 374, 857–863 (2021). https://doi.org/10.1126/science.abj9195


