Abstract
Claudin family proteins play an important role in the formation of tight junctions in several tissues. Individual claudins display organ- and tissue-specific expression. Claudin domain containing 1 (CLDND1), also known as claudin 25 (CLDN25), is a homolog of the claudin family, and its expression was reported to be downregulated in a mouse model of cerebellar hemorrhage. We have also reported that the retinoic acid receptor-related orphan receptor α (RORα) is involved in the transcriptional activation of CLDND1 by binding to the RORα responsive element (RORE) in the CLDND1 promoter region. Cholesterol and its derivative oxysterol reportedly serve as ligands for the nuclear receptor RORα. However, the effect of cholesterols on CLDND1 expression is unclear. The present study aimed to evaluate the effect of inhibiting steroid synthesis via lovastatin on RORα-mediated CLDND1 transcriptional regulation. Chromatin immunoprecipitation and luciferase reporter assays revealed that RORα-mediated transcriptional regulation of CLDND1 was suppressed upon lovastatin treatment of HepG2 cells; however, this inhibitory effect was attenuated by supplementation with cholesterol. Furthermore, quantitative reverse transcription-PCR and immunoblotting analyses revealed the downregulated expression of CLDND1 mRNA and protein in HepG2 cells upon lovastatin treatment with no parallel changes in RORα mRNA and protein levels. These results confirm that cholesterol serve as ligands for RORα and are, therefore, involved in the activation of CLDND1 transcriptional regulation by RORα.
INTRODUCTION
Increased permeability of the endothelial cells lining the blood vessels in the brain causes vascular edema, which can ultimately result in a stroke. Vascular permeability is regulated by tight junctions (TJs) comprising claudins as the main components, along with occludin proteins. Twenty-seven claudins have been identified in humans to date1); they are differentially expressed depending on the cell type and pathological condition.2) Claudin domain containing 1 (CLDND1), also known as claudin25 (CLDN25), is highly expressed in the cerebellum and one of the target genes of the retinoic acid receptor-related orphan receptor α (RORα).
Staggerer (sg/sg) mutant mice carry a deletion within the RORα receptor gene, exhibit excessive inflammation and lipid metabolism abnormalities and enhanced macrophage deposition in the vascular walls, and also develop severe atherosclerosis.3,4) RORα regulates the target gene transcription by binding as a monomer to a RORα response element (RORE), which contains a consensus motif of RGGTCA (R; A or G) preceded by an A/T-rich sequence.5) Many ROREs have been found within gene promoters, but only a few genes have been functionally demonstrated to transcriptional regulated by RORα. Cholesterol and its derivative oxysterol serve as ligands for RORα.6,7) Synthetic RORα ligands may be used as drug targets for the treatment of several diseases such as atherosclerosis8,9) and non-alcoholic steatohepatitis.10)
We previously reported that RORα is involved in the transcriptional activation of CLDND1 by binding to a RORE present in the CLDND1 promoter region,11) and the microRNA miR-124 plays a role in the post-transcriptional regulation of CLDND1.12) Moreover, the mRNA expression levels of CLDND1 and RORα correlate with each other in rat tissues.11) A previous study using a mouse model of cerebellar hemorrhage reported that the expression of CLDND1 is downregulated during cerebellar hemorrhage, and CLDND1 knockdown in human vascular endothelial cells resulted in an enhanced vascular permeability.13) Therefore, we hypothesized that insufficient transcriptional regulation of CLDND1 by RORα is associated with the pathogenesis of cerebrovascular conditions such as cerebral hemorrhage. However, the transcriptional regulation of CLDND1 has not been studied extensively to date, and the effect of cholesterol on the RORα-mediated transcriptional regulation of CLDND1 remains unknown.
As cholesterol-lowering agents, statins have proven to be effective in preventing the onset of stroke by inhibiting 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme in the cholesterol biosynthesis pathway.14,15) However, as a scarcely reported side effect of statins, the reduction in total cholesterol has been associated with an increased risk of hemorrhagic stroke.16,17) In this study, we used lovastatin to evaluate the effect of cholesterol on RORα-mediated CLDND1 transcriptional regulation in vitro in the human liver carcinoma cell line HepG2.
MATERIALS AND METHODS
Cell Culture
Immortalized HepG2 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 100 μg/mL penicillin-streptomycin and 10% fetal bovine serum (FBS) at 5% CO2 and 37°C.
Chromatin Immunoprecipitation Assay (ChIP)
HepG2 cells were seeded in 90 mm dishes and grown to 90% confluence (24 h) in DMEM containing 10% FBS before lovastatin treatment. The cells were grown for another 72 h in fresh media. At that time, 10 μM lovastatin or DMSO was added to the cells. For cells to which lovastatin treated, a medium without FBS was used. ChIP assay was conducted following the manufacturer’s instructions for the OneDay ChIP kit (Diagenode, Liege, Belgium). The reactions were carried out overnight at 4°C, with mixing, using 1 μg of either anti-RORα (C-16) antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or normal nonimmunized IgG as a negative control. Sample DNA was purified and amplified by PCR using designed primer specific (hCLDND1-RORE_FW: 5’-GGATACTGTCTGCCAGGTCAC-3’, hCLDND1-RORE_RV: 5’-CGGTCATCTCGAGTCAATGAC-3’) to RORα promoter region. PCR products were separated in a 3% agarose gel and visualized using ethidium bromide and a CS Analyzer (Atto, Tokyo, Japan).
Luciferase Reporter Constructs
The luciferase reporter plasmid pRORE×3 was constructed as follows. Synthetic oligonucleotides corresponding to CLDND1-RORE-sense (5’-GGAAATAAAATGGGTCAACGTT-3’) and CLDND1-RORE-antisense (5’-AACGTTGACCCATTTTATTTCC-3’) containing a RORE of CLDND1 genes were phosphorylated with the T4 DNA polynucleotide kinase (Takara Bio, Shiga, Japan), then mixed and annealed. The resulting double-stranded oligonucleotide was cloned into the SmaI site of the luciferase expression reporter vector PGVP2 (Nippon Gene, Tokyo, Japan) containing an SV40 promoter. Then, the plasmid clone containing the three sense-oriented tandem copies of RORE (pRORE×3) was selected by sequencing of the resulting recombinant DNA clones.
Transfection and Luciferase Activity Assay
HepG2 cells were transfected using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Cells were seeded at 0.5×105 cells/well in DMEM. After 9 h at 37°C and 5% CO2, the cells in each well were transfected for 12 h-16 h with a mixture of 100 ng of the luciferase reporter plasmid (pRORE×3), 100 ng of the RORα expression plasmid (pRORα) or empty plasmid (pSG5), and 100 ng of the β-galactosidase reporter plasmid, which was used to normalize the luciferase activity. The cells were grown for another 24 h-32 h in fresh media. At that time, 20 μM lovastatin (Merck, Tokyo, Japan) or DMSO was added to the cells. In some cases, 50 nM cholesterol was added with lovastatin. Luciferase activity was determined using the Picagene luciferase assay kit (Toyo Ink, Tokyo, Japan) according to the manufacturer’s protocol, and measured with a Luminescencer-PSN AB-2200 (Atto).
Quantitative Reverse Transcription-PCR (qRT-PCR)
HepG2 cells were seeded at 1×105cells/well in DMEM. After 24 h at 37°C and 5% CO2, 20 μM lovastatin or DMSO were added to the cells, which were cultured in DMEM supplemented 10% fetal bovine serum. After 24 h at 37°C and 5% CO2, the cells were harvested by using ISOGEN reagents (Nippon Gene). Total RNA was prepared using ISOGEN according to the manufacturer’s protocol and reverse-transcribed for 90 min at 37°C in a reaction using 200 U Moloney murine leukemia virus reverse transcriptase (Thermo Fisher Scientific). The cDNA was directly amplified using a Roche Light Cycler in a 12 μL reaction containing SYBR Green Real-Time PCR master mix (Toyobo, Osaka, Japan) and 1 μM primers. The primers used to amplify the selected gene have been shown in Table 1. Gene expression was quantified using the Light Cycler software (Roche, Basel, Switzerland) as the second derivative maximum of the curve.
Table 1. Real-Time PCR Primers Sequences
| Primer |
Sequence |
| rtCLDND1-FW |
5’- TAACTGAGCAGTTCATGGAG -3’ |
| rtCLDND1-RV |
5’- TAAGCTTCGGCAAATGCAAG -3’ |
| rtRORA-FW |
5’- TCCATGCAAGATCTGTGGAG -3’ |
| rtRORA-RV |
5’- ACAGCATCTCGAGACATCCC -3’ |
| rtMPD-FW |
5’- CTCTTACCTCAATGCCATCTC -3’ |
| rtMPD-RV |
5’- CACAAACTCAGCCACAGTGTC -3’ |
| rt18SrRNA-FW |
5’- CGATAACGAACGAGACTCTGG -3’ |
| rt18SrRNA-RV |
5’- TAGGGTAGGCACACGCTGAGC -3’ |
Immunoblotting
HepG2 cells were seeded at 5×105cells/well in DMEM. After an incubation of 24 h at 37°C and 5% CO2, 20 μM lovastatin or DMSO was added to the cells. The cells were grown for another 72 h in fresh media. The cells were homogenized in chilled lysis buffer containing 50 mM Tris-HCl (pH 7.0), 200 mM sucrose, 1 mM ethylenediaminetetraacetic acid, 1 mg/mL leupeptin, 1 mg/mL pepstatin, 0.5 mM phenylmethylsulfonyl fluoride, and 1% sodium dodecyl sulfate (SDS).The samples were subjected to 12% SDS polyacrylamide gel electrophoresis (PAGE), and followed by transfer to an Immobilon-P membrane (Merck Millipore, Billerica, MA). The membranes were incubated overnight at 4°C with anti-CLDND1 (a custom antibody against the epitope in the first loop peptide region, produced by Eurofins Genomics [13054V;Tokyo,Japan]) or with anti-RORα H-65 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or with anti-β-actin (Proteintech Group, Chicago, USA) antibodies. Subsequently, the membranes were incubated with ECL anti-rabbit or mouse IgG, horseradish peroxidase-linked whole antibody (GE Healthcare, Buckinghamshire, UK). The membranes were detected with the ECL Western blotting detection reagents (GE Healthcare) according to the manufacturer’s instructions. The band intensities were analyzed with a CS Analyzer (Atto).
Statistical Analysis
All experiments were performed at least three times. Data have been expressed as the mean ± SD unless otherwise specified. Error bars represent the mean ± S.E. Comparisons of two were performed with the Student’s t-test. Differences between groups were evaluated by one-way ANOVA. When the F value was significant (p < 0.05), the Tukey-Kramer test was performed for post-hoc comparison. Values were considered to be statistically significant when p <0.05.
RESULTS
Binding of RORα to RORE Present in the CLDND1 Promoter Region is Suppressed by Lovastatin
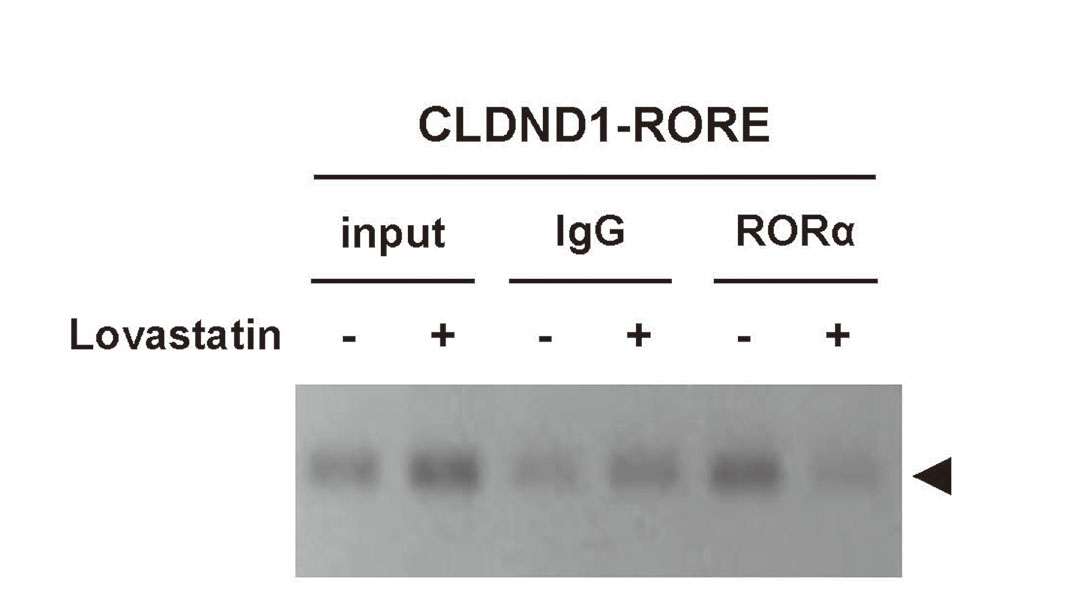
ChIP-PCR assay was performed to investigate the effect of lovastatin on the binding of RORα to RORE present in the CLDND1 promoter region. The chromatin fragments of the CLDND1 promoter region, including RORE, without lovastatin treatment, were immunoprecipitated by the anti-RORα antibody. As a control, the addition of a non-immune IgG antibody immunoprecipitated a small amount of the chromatin fragment of the CLDND1 promoter. In contrast, the amount of immunoprecipitation of the CLDND1 promoter chromatin region containing RORE after lovastatin treatment was lower than that of the vehicle. The amount of immunoprecipitation with the anti-RORα antibody after lovastatin treatment was lower than that with the non-immune IgG antibody (Fig. 1).
Cholesterol Restores the RORα-Mediated CLDND1 Transcription Reduced by Lovastatin
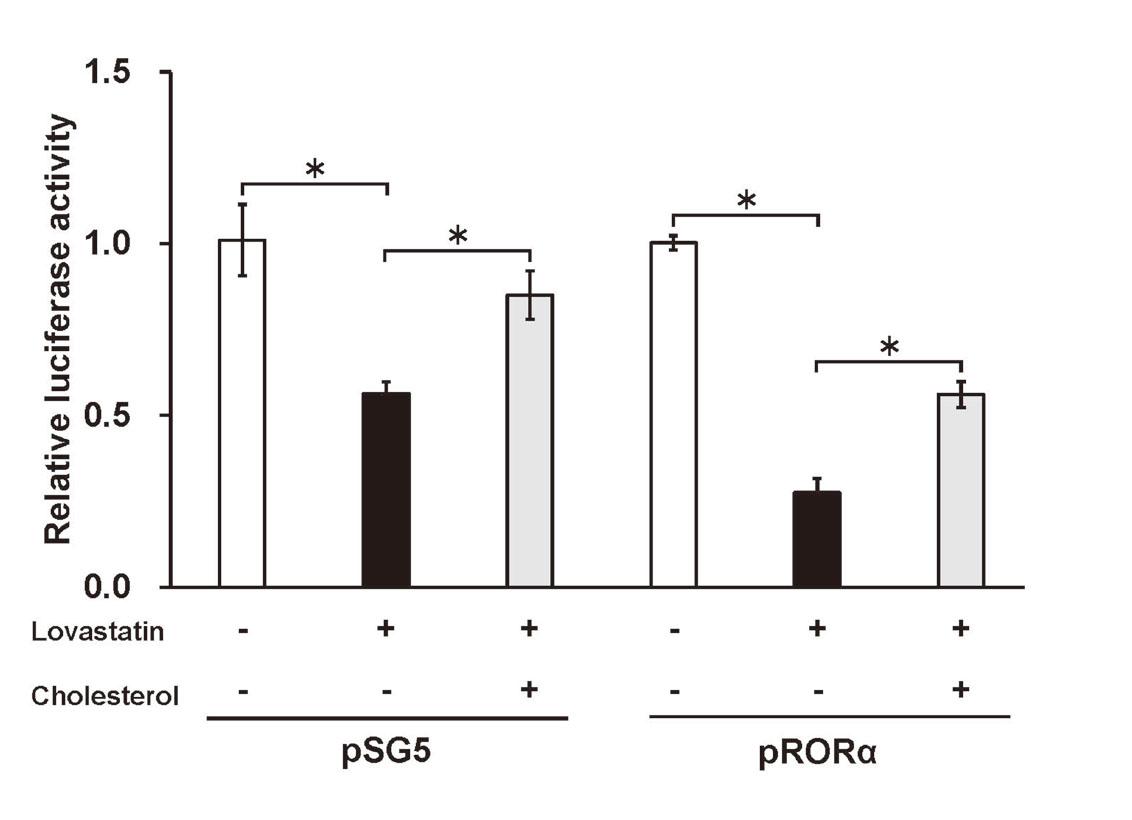
We conducted a luciferase reporter assay to assess the effect of lovastatin on the RORα-mediated CLDND1 transcription. For this reason, HepG2 cells were co-transfected with an empty expression plasmid (pSG5) and the luciferase reporter plasmid containing three copies of the RORE (pROREx3) or with the RORα expression plasmid (pRORα) and pROREx3; each group was treated or untreated with lovastatin. Additionally, we evaluated the effect of lovastatin with and without cholesterol. The results showed enhanced luciferase activity after co-transfection with the pRORα and pROREx3 plasmids; however, upon the addition of lovastatin, the luciferase activity was significantly suppressed. We observed similar results following the transfection of HepG2 cells with the pROREx3 plasmid alone. The luciferase activity was significantly increased upon the addition of cholesterol with lovastatin. Moreover, the addition of cholesterol restored the increased luciferase activity induced by endogenous RORα (i.e., with pROREx3 alone) and RORα expression (i.e., with the pROREx3 and RORα expression plasmids) (Fig. 2).
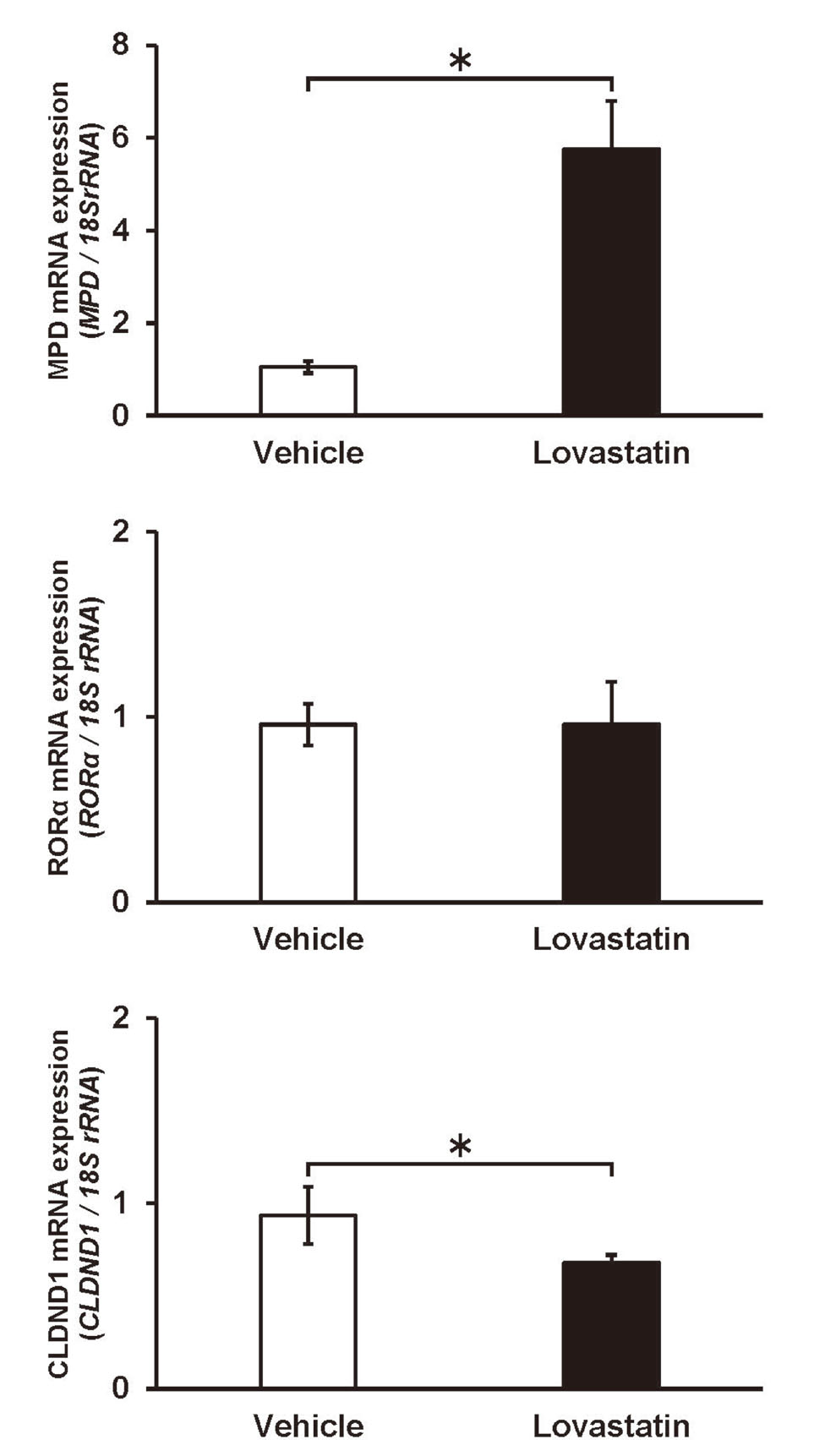
We performed qRT-PCR to evaluate the effect of lovastatin treatment on CLDND1 mRNA expression levels. RNA was prepared 24 h after adding lovastatin to the HepG2 cells, and we evaluated the mRNA expression levels of mevalonate pyrophosphate decarboxylase (MPD), RORα, and CLDND1. As statin treatment is known to enhance the expression of MPD in the liver,18) the expression level of MPD was evaluated to confirm the action of lovastatin as a positive control. Following lovastatin treatment, the expression level of MPD was about 6-fold higher than that in the vehicle group, confirming the action of lovastatin on HepG2 cells. The results also showed that lovastatin treatment reduced the CLDND1 mRNA expression level by 27.6% compared to that in the vehicle group. In contrast, there was no significant difference in RORα mRNA expression between lovastatin- and vehicle-treated cells (Fig. 3).
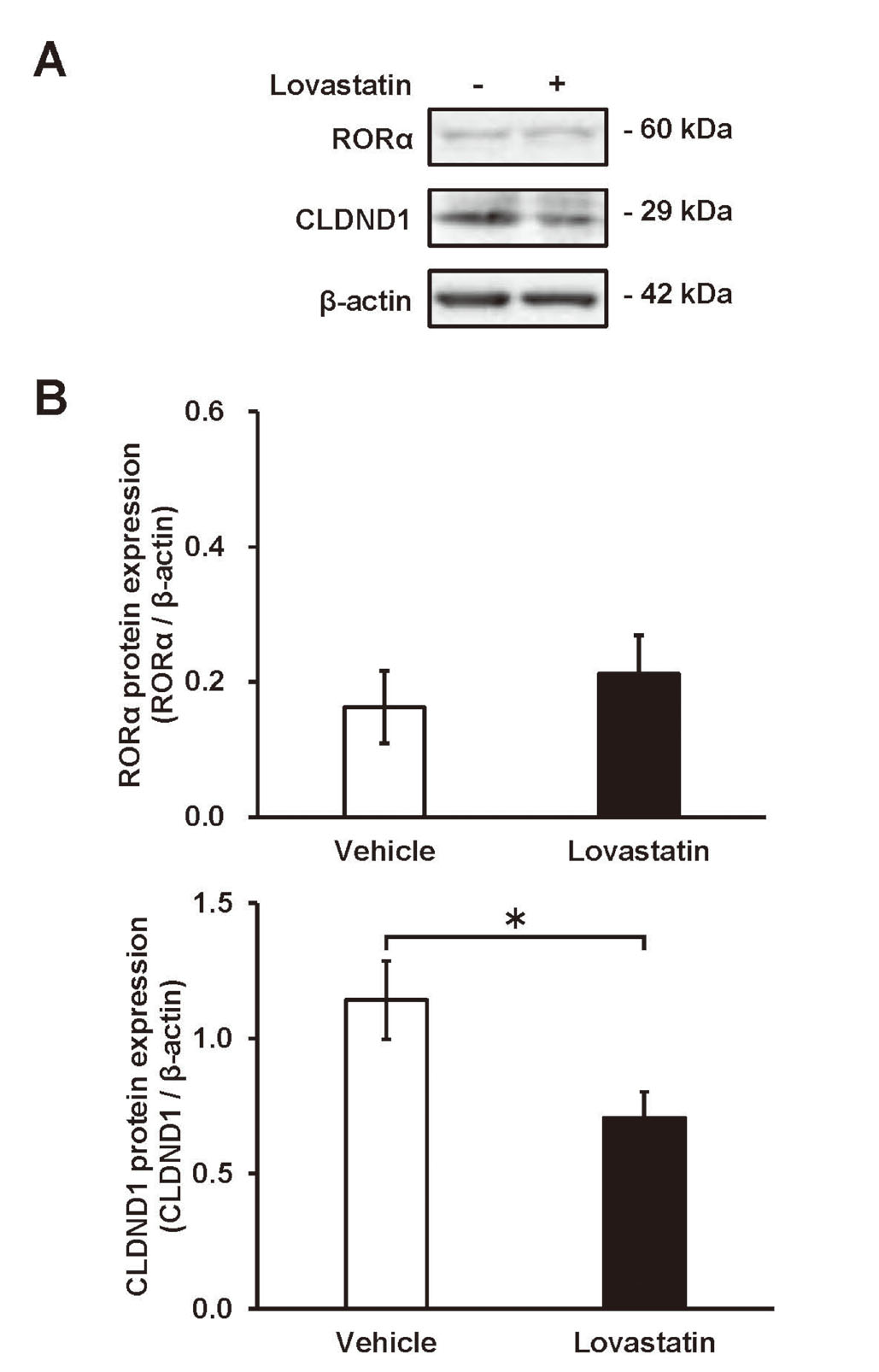
Immunoblotting was performed to evaluate the effect of lovastatin treatment on the CLDND1 protein expression. Proteins were prepared 72 h after the addition of lovastatin to the HepG2 cells, and the protein expression levels of RORα and CLDND1 were measured. As with the expression of CLDND1 mRNA, lovastatin treatment significantly reduced the CLDND1 protein expression, and the protein level was 24.4% lower than that in the vehicle group. However, the protein expression of RORα (like RORα mRNA expression) was not significantly altered by lovastatin treatment (Fig. 4).
DISCUSSION
RORα has been reported to activate CLDND1 transcription by binding to the RORE present in the CLDND1 promoter region.11) The present study demonstrates that lovastatin inhibits this RORα-mediated CLDND1 transcription. ChIP assay showed that the binding of RORα to the RORE present in the promoter region of CLDND1 was suppressed in HepG2 cells by lovastatin treatment. Furthermore, luciferase reporter assays revealed that lovastatin affected RORα-mediated CLDND1 transcription. These results suggest that the inhibition of the synthesis of steroid analogs by lovastatin treatment reduces the binding between RORE and RORα, thereby suppressing RORα-activated CLDND1 transcription. The inhibition of RORα responsiveness by lovastatin treatment was abrogated by cholesterol supplementation, indicating the direct involvement of cholesterol in the RORα-mediated transcription regulation. However, RORα responsiveness was not completely restored and still reduced relative to that of the untreated control group. Therefore, intermediate metabolites other than cholesterol and cholesterol metabolites inhibited by lovastatin may also be involved in the activation of transcription by RORα.
RORα has been considered an orphan receptor with no known ligand; however, recent studies identified cholesterol as a ligand for RORα.6,7) Although cholesterol derivatives (e.g., 7-oxygenated sterols and 24S-hydroxycholesterol) reportedly function as RORα inverse agonists and regulate RORα target genes,19,20) their effects on the RORα target gene CLDND1 remained unknown. Some studies have suggested that modulation of the RORα target by synthetic ligands may warrant cholesterol supplementation.8,21)
Furthermore, we observed significant decreases in the mRNA and protein expression of CLDND1 upon lovastatin treatment without any corresponding change in the protein and mRNA expression of RORα. Thus, it is suggested that steroid analogs do not regulate CLDND1 transcription by regulating RORα expression but rather by improving the binding affinity between RORα and the RORE in the CLDND1 promoter region.
Stroke accounts for the majority of cerebrovascular diseases. Hypertension directly contributes to the development of strokes,22) and the degeneration of small arteries, necrosis, and formation of small aneurysms in the brain are known to be the major causes of vascular rupture and thrombus formation.23) Therefore, increased vascular permeability is an important factor in the development of stroke. TJs control the vascular permeability, a risk factor for cerebral hemorrhage.24) CLDND1 is one of the main constituent molecules of TJs and highly expressed in the cerebellum. The expression of CLDND1 and RORα is positively correlated in rat tissues and robust in the cerebellum and liver.11) CLDND1 expression was reduced in a mouse model of cerebellar hemorrhage, and knockdown of CLDND1 in human brain endothelial cells resulted in an enhanced vascular permeability.13) These results suggest that decreased expression of CLDND1 causes cerebral hemorrhage in the cerebellum. As most of the cholesterol is synthesized in the liver in vivo, liver cells are considered to be less affected by lovastatin than other cell types. However, the inhibition of cholesterol synthesis in HepG2 cells by lovastatin treatment significantly reduced the expression level of CLDND1. Similarly, lower cholesterol levels may reduce RORα-mediated CLDND1 expression in other cells.
Epidemiological data indicate that stroke-related risk factors include changes in cholesterol levels.25,26) Furthermore, a reduction in the total cholesterol level was associated with an increased risk of hemorrhagic stroke.27) Hence, lowering cholesterol levels with a statin may cause cerebral hemorrhage. Cholesterol reduction may weaken cell membranes and lead to the collapse of TJs owing to the suppression of the transcription of cell adhesion molecules.
Stroke-prone spontaneously hypertensive rats (SHRSP) are used as a stroke model because they exhibit severe hypertension and frequently suffer strokes.28) SHRSP exhibit hemorrhage in the cerebrum but rarely in the cerebellum.29) The serum cholesterol level of SHRSP is lower than that of normotensive Wistar Kyoto rats (WKY).30) Furthermore, it has been reported that the cholesterol content is decreased in most parts of the brain; however, it is increased in the cerebellum of SHRSP compared to that of WKY.31) It was also reported that the expression level of CLDND1 was not changed in the cerebrum but increased in the cerebellum of SHRSP as compared with that in WKY.12) These results show that in SHRSP, cholesterol might promote RORα-mediated expression of CLDND1 and forms TJs to suppress cerebellar hemorrhage.
In conclusion, we found that cholesterol enhances CLDND1 transcription regulation by improving the binding affinity between RORα and RORE in the CLDND1 promoter region. Thus, the administration of lovastatin to lower cholesterol levels should be used with caution in clinical settings as the statin may also increase the risk of a cerebral hemorrhage in the cerebellum.
Acknowledgments
We thank Aki Tamura and Sou Kobayashi from Fukuyama University for technical assistance. This work was supported by Fukuyama University Grant for Academic Research Projects (grant no. GARP 2019-103).
Conflict of interest
The authors declare no conflict of interest.
REFERENCES
- 1) Mineta K, Yamamoto Y, Yamazaki Y, Tanaka H, Tada Y, Saito K, Tamura A, Igarashi M, Endo T, Takeuchi K, Tsukita S. Predicted expansion of the claudin multigene family. FEBS Lett., 585, 606–612 (2011).
- 2) Lal-Nag M, Morin PJ. The claudins. Genome Biol., 10, 235 (2009).
- 3) Jarvis CI, Staels B, Brugg B, Lemaigre-Dubreuil Y, Tedgui A, Mariani J. Age-related phenotypes in the staggerer mouse expand the RORα nuclear receptor’s role beyond the cerebellum. Mol. Cell. Endocrinol., 186, 1–5 (2002).
- 4) Mamontova A, Séguret-Macé S, Esposito B, Chaniale C, Bouly M, Delhaye-Bouchaud N, Luc G, Staels B, Duverger N, Mariani J, Tedgui A. Severe atherosclerosis and hypoalphalipoproteinemia in the staggerer mouse, a mutant of the nuclear receptor RORα. Circulation, 98, 2738–2743 (1998).
- 5) Giguere V, Tini M, Flock G, Ong E, Evans RM, Otulakowski G. Isoform-specific amino-terminal domains dictate DNA-binding properties of RORα, a novel family of orphan hormone nuclear receptors. Genes Dev., 8, 538–553 (1994).
- 6) Kallen JA, Schlaeppi JM, Bitsch F, Geisse S, Geiser M, Delhon I, Fournier B. X-ray structure of the hRORα LBD at 1.63 Å: structural and functional data that cholesterol or a cholesterol derivative is the natural ligand of RORα. Structure, 10, 1697–1707 (2002).
- 7) Kallen J, Schlaeppi JM, Bitsch F, Delhon I, Fournier B. Crystal Structure of the Human RORα Ligand Binding Domain in Complex with Cholesterol Sulfate at 2.2 Å. J. Biol. Chem., 279, 14033–14038 (2004).
- 8) Billon C, Sitaula S, Burris TP. Inhibition of RORα/γ suppresses atherosclerosis via inhibition of both cholesterol absorption and inflammation. Mol. Metab., 5, 997–1005 (2016).
- 9) Matsuoka H, Tokunaga R, Katayama M, Hosoda Y, Miya K, Sumi K, Ohishi A, Kamishikiryo J, Shima A, Michihara A. Retinoic acid receptor-related orphan receptor α reduces lipid droplets by upregulating neutral cholesterol ester hydrolase 1 in macrophages. BMC Mol. Cell Biol., 21, (2020).
- 10) Han YH, Kim HJ, Na H, Nam MW, Kim JY, Kim JS, Koo SH, Lee MO. RORα Induces KLF4-Mediated M2 Polarization in the Liver Macrophages that Protect against Nonalcoholic Steatohepatitis. Cell Rep., 20, 124–135 (2017).
- 11) Matsuoka H, Shima A, Uda A, Ezaki H, Michihara A. The retinoic acid receptor-related orphan receptor a positively regulates tight junction protein claudin domain-containing 1 mRNA expression in human brain endothelial cells. J. Biochem., 161, 441–450 (2017).
- 12) Matsuoka H, Tamura A, Kinehara M, Shima A, Uda A, Tahara H, Michihara A. Levels of tight junction protein CLDND1 are regulated by microRNA-124 in the cerebellum of stroke-prone spontaneously hypertensive rats. Biochem. Biophys. Res. Commun., 498, 817–823 (2018).
- 13) Ohnishi M, Ochiai H, Matsuoka K, Akagi M, Nakayama Y, Shima A, Uda A, Matsuoka H, Kamishikiryo J, Michihara A, Inoue A. Claudin domain containing 1 contributing to endothelial cell adhesion decreases in presence of cerebellar hemorrhage. J. Neurosci. Res., 95, 2051–2058 (2017).
- 14) Di Mascio R, Marchioli R, Tognoni G. Cholesterol reduction and stroke occurrence: an overview of randomized clinical trials. Cerebrovasc. Dis., 10, 85–92 (2000).
- 15) Bucher HC, Griffith LE, Guyatt GH. Effect of HMGcoA reductase inhibitors on stroke: A meta-analysis of randomized, controlled trials. Ann. Intern. Med., 128, 89–95 (1998).
- 16) Goldstein LB, Amarenco P, Szarek M, Callahan A, Hennerici M, Sillesen H, Zivin JA, Welch KM. Hemorrhagic stroke in the Stroke Prevention by Aggressive Reduction in Cholesterol Levels study. Neurology, 70, 2364–2370 (2008).
- 17) Tirschwell DL, Smith NL, Heckbert SR, Lemaitre RN, Longstreth WT, Psaty BM. Association of cholesterol with stroke risk varies in stroke subtypes and patient subgroups. Neurology, 63, 1868–1875 (2004).
- 18) Michihara A, Akasaki K, Yamori Y, Tsuji H. Change in the protein level of mevalonate pyrophosphate decarboxylase in tissues of mouse by pravastatin. Biol. Pharm. Bull., 26, 1082–1085 (2003).
- 19) Wang Y, Kumar N, Solt LA, Richardson TI, Helvering LM, Crumbley C, Garcia-Ordonez RD, Stayrook KR, Zhang X, Novick S, Chalmers MJ, Griffin PR, Burris TP. Modulation of retinoic acid receptor-related orphan receptor α and γ activity by 7-oxygenated sterol ligands. J. Biol. Chem., 285, 5013–5025 (2010).
- 20) Wang Y, Kumar N, Crumbley C, Griffin PR, Burris TP. A second class of nuclear receptors for oxysterols: Regulation of RORα and RORγ activity by 24S-hydroxycholesterol (cerebrosterol). Biochim. Biophys. Acta Mol. Cell Biol. Lipids, 1801, 917–923 (2010).
- 21) Kojetin DJ, Burris TP. REV-ERB and ROR nuclear receptors as drug targets. Nat. Rev. Drug Discov., 13, 197–216 (2014).
- 22) MacMahon S, Peto R, Collins R, Godwin J, MacMahon S, Cutler J, Sorlie P, Abbott R, Collins R, Neaton J, Abbott R, Dyer A, Stamler J. Blood pressure, stroke, and coronary heart disease. Part 1, prolonged differences in blood pressure: prospective observational studies corrected for the regression dilution bias. Lancet, 335, 765–774 (1990).
- 23) Hademenos GJ, Massoud TF. Biophysical mechanisms of stroke. Stroke, 28, 2067–2077 (1997).
- 24) Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol., 2, 285–293 (2001).
- 25) Tanaka H, Ueda Y, Hayashi M, Date C, Baba T, Yamashita H, Shoji H, Tanaka Y, Owada K, Detels R. Risk factors for cerebral hemorrhage and cerebral infarction in a japanese rural community. Stroke, 13, 62–73 (1982).
- 26) Benfante R, Yano K, Hwang LJ, Curb JD, Kagan A, Ross W. Elevated serum cholesterol is a risk factor for both coronary heart disease and thromboembolic stroke in hawaiian japanese men implications of shared risk. Stroke, 25, 814–820 (1994).
- 27) Yano K, Reed DM, MacLean CJ. Serum cholesterol and hemorrhagic stroke in the honolulu heart program. Stroke, 20, 1460–1465 (1989).
- 28) Yamori Y. Predictive and Preventive Pathology of Cardiovascular Diseases. Pathol. Int., 39, 683–705 (1989).
- 29) K. O. Y. Y. A. N. Establishment of the stroke prone spontaneously hypertensive rat (SHR). Circ. Res., 34/35, 143–153 (1974).
- 30) Iritani N, Fukuda E, Nara Y, Yamori Y. Lipid metabolism in spontaneously hypertensive rats (SHR). Atherosclerosis, 28, 217–222 (1977).
- 31) Katayama M, Matsuoka H, Hamashima T, Michihara A. Brain Reg. Reduc. Amounts Mevalonate Pyrophosphate Decarboxylase Correspond Sites Strokes Stroke-Prone Spontaneously Hypertensive Rats., 3, 106–112 (2020).