要旨
症例は25歳女性,12歳から横紋筋融解症を繰り返していた.妊娠悪阻による飢餓を契機に横紋筋融解症が再燃し,当科紹介となった.血液検査でCK値の上昇と,総カルニチンの低下を認めた.末梢血リンパ球のpalmitoyl-CoA dehydrogenase活性が低値であり,極長鎖アシルCoA脱水素酵素(very-long-chain acyl-coenzyme A dehydrogenase,以下VLCADと略記)欠損症と診断した.ACADVL遺伝子のc.1349G>A(p.R450H)変異に加え,c.1332G>A変異との複合ヘテロ変異を認めた.VLCAD欠損症は脂肪酸酸化異常症の一つで,横紋筋融解症を生じうる.未診断のVLCAD欠損症が妊娠悪阻を契機に診断に至ることがある.
Abstract
A 25-year-old Japanese woman with a history of repeated episodes of rhabdomyolysis since the age of 12 presented with rhabdomyolysis caused by hyperemesis gravidarum. Blood tests showed an elevated serum CK level (11,755 IU/l; normal: 30–180 IU/l). Carnitine fractionation analysis revealed low levels of total carnitine (18.3 μmol/l; normal: 45–91 μmol/l), free carnitine (13.1 μmol/l; normal: 36–74 μmol/l), and acylcarnitine (5.2 μmol/l; normal: 6–23 μmol/l). Tandem mass spectrometry showed high levels of C14:1 acylcarnitine (0.84 nmol/ml: normal: <0.4 nmol/ml) and a high C14:1/C2 ratio of 0.253 (normal: <0.013), indicating a potential diagnosis of very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency. Enzyme activity measurement in the patient’s peripheral blood lymphocytes confirmed the diagnosis of VLCAD deficiency, with low palmitoyl-CoA dehydrogenase levels (6.5% of normal control value). With the patient’s informed consent, acyl-CoA dehydrogenase very long-chain (ACADVL) gene analysis revealed compound heterozygous mutations of c.1332G>A in exon 13 and c.1349G>A (p.R450H) in exon 14. In Japan, neonatal mass screening is performed to detect congenital metabolic diseases. With the introduction of tandem mass screening in 2014, fatty acid metabolism disorders, including VLCAD deficiency, are being detected before the onset of symptoms. However, it is important to note that mass screening cannot detect all cases of this disease. For patients with recurrent rhabdomyolysis, it is essential to consider congenital diseases, including fatty acid metabolism disorders, as a potential diagnosis.
はじめに
極長鎖アシルCoA脱水素酵素(very-long-chain acyl-coenzyme A dehydrogenase,以下VLCADと略記)は,長期間の飢餓状態や,長時間の運動の際などに,脂肪酸からエネルギーを合成するのに必須の酵素であり1),ミトコンドリアの脂肪酸β酸化に関与する2).VLCAD欠損症は常染色体潜性遺伝形式をとる稀な遺伝性疾患であり,長鎖脂肪酸の代謝障害により,長時間の絶食や運動・労作を誘因として,低ケトン性低血糖や心筋症,横紋筋融解症などの急性の代謝障害を生じる3).VLCAD欠損症は,発症前型,新生児発症型,乳幼児期発症型(肝型),遅発型(骨格筋型)の四つの臨床病型に分かれる4).脳神経内科で主に診療するのは遅発型であり,学童期以降に横紋筋融解やミオパチーなどで発症することが多い.わが国では先天性代謝異常の新生児マススクリーニング対象疾患5)で,その頻度は93,000人に1人程度とされている6)が,成人期に診断される例は頻度が低く,発作間欠期には血清アシルカルニチン分析でも特徴的な所見を認めない場合があり,診断に難渋することがある7).我々は,妊娠悪阻による長期の飢餓状態を契機に横紋筋融解症を来し,アシルカルニチン分析や脂肪酸代謝酵素の活性評価からVLCAD欠損症と診断した1症例を経験した.本症例は複合ヘテロ変異を有していたこと,反復性横紋筋融解症の鑑別診断としてVLCAD欠損症をはじめとする脂肪酸代謝異常症を鑑別に挙げる必要があること,妊娠悪阻による飢餓状態でVLCAD欠損症が悪化しうることなど,示唆に富む症例であったため報告する.
症例
患者:25歳女性
主訴:歩行困難,筋肉痛
既往歴:1歳4ヶ月時に川崎病の既往がある.
家族歴:父に運動後に生じる下肢痛と褐色尿の既往があるが,遺伝子検査は施行していない.同胞2人には類症なし.
生活歴:飲酒なし,喫煙なし.
現病歴:出生発育に問題なく,過去の健診で検尿の異常は指摘されなかった.12歳時から,激しい運動をした後に下肢の筋痛と褐色尿を自覚していたが,安静により改善していたため放置していた.15歳時,連日の運動の後,歩行困難が出現して他院に入院した際に,CKが92,300 IU/lと著明に高値であり,横紋筋融解症と診断された.その後は過度の運動を控えるようになり,下肢の筋痛や褐色尿の頻度は減少していた.25歳時に初めて妊娠した際,悪阻により500 kcal/日程度しか摂取できない日が約5日続いた後から,下肢の筋痛が出現し歩行困難となり,他院に搬送された.その際にCK値が11,755 IU/lと著明に上昇していたため,横紋筋融解症の再燃と診断された.原因精査を目的に当科へ入院となった.
入院時所見:身長162.2 cm,体重45.5 kgでやせ型である.血圧87/37 mmHg,心拍77回/分.一般身体所見に特記事項はない.神経学的所見では脳神経に嚥下障害,構音障害を含めて特記所見は見られなかった.運動系では四肢や体幹の明らかな筋萎縮や肥大はみられず,握力は右19 kg,左16 kgであった.徒手筋力テストでは四肢筋力低下はないが,両下肢の近位筋に把握痛を認めた.入院時は起立,歩行は正常となっており,その他の神経学的所見に異常はなかった.
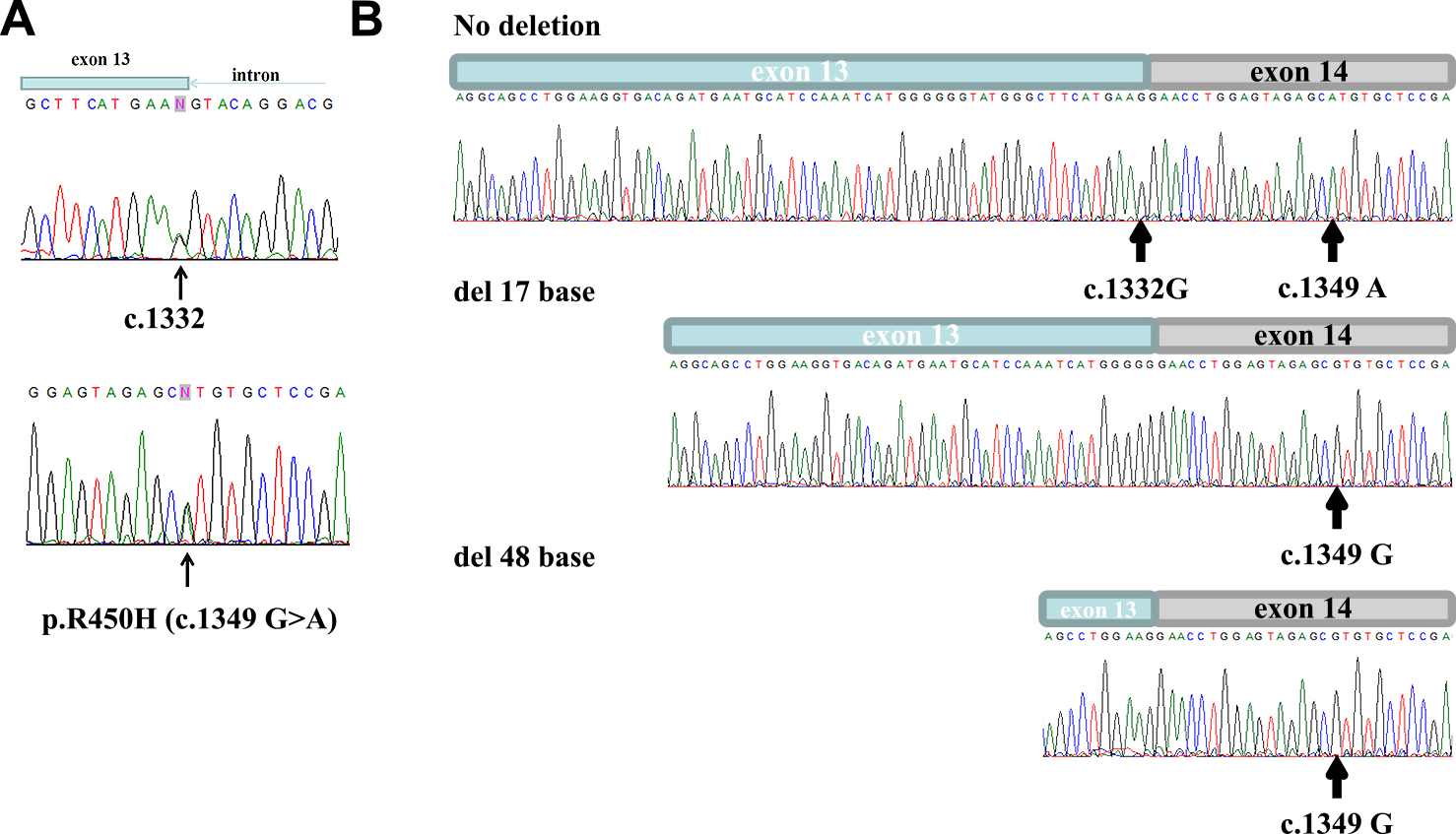
入院時検査所見:血液所見では,血清CK 237 IU/l(正常値30~180 IU/l)と軽度高値であった.カルニチン分画では総カルニチン18.3 μmol/l(正常値45~91 μmol/l),遊離カルニチン13.1 μmol/l(正常値36~74 μmol/l),アシルカルニチン5.2 μmol/l(正常値6~23 μmol/l)といずれも低値であった.タンデム質量分析ではC14:1アシルカルニチンが0.84 nmol/ml(正常値 <0.4 nmol/ml),C14:1/C2比0.253(カットオフ <0.013)と,ともに高値であった(Table 1).電解質,甲状腺機能,自己抗体,乳酸,ピルビン酸,アンモニアはいずれも基準範囲内であった.一般尿検査ではミオグロビン尿などの異常所見は指摘できなかった.尿中有機酸分析では異常所見を認めなかった.針筋電図検査では左三角筋,左上腕二頭筋にて弱収縮時に低振幅・短持続時間の運動単位電位を認めた.経胸壁心臓超音波検査では左室駆出率は70%で,壁運動異常もなく,心機能は保たれていた.呼吸機能検査では肺活量2,140 ml,%肺活量68%と軽度の拘束性障害の所見であった.筋生検は施行していない.血中カルニチン分画の低下に加えて,タンデム質量分析でC14:1アシルカルニチンとC14:1/C2比の上昇が判明したためVLCAD欠損症を疑った.患者末梢血リンパ球のpalmitoyl-CoA dehydrogenase活性を測定したところ,正常者平均の6.5%と低値であったため,VLCAD欠損症の診断に至った.倫理審査を経て,本人から文書での同意を得てACADVL遺伝子解析を行ったところ,exon 13にc.1332G>Aとexon 14にはc.1349G>A (p.R450H)という2種類の1塩基置換を認めた(Fig. 1A).c.1349G>Aは,複数の骨格筋型VLCAD欠損症例の病因として既報の変異であり8),c.1332G>Aは心筋症を伴う新生児期発症のホモ接合性患者が報告されている9).後者はアミノ酸が変化しない同義置換だが,exon 13の3'末端の塩基であり,スプライシングへの影響が推測された.その検証として,患者白血球から抽出したRNAからrandom primerを用いてtotal cDNAを合成し,ACADVL exon 11~15をPCR増幅・精製し,これをベクターに組み込んで大腸菌に導入した.培養後,得られた40クローンについてmRNAの塩基配列を解析した結果,24クローンがc.1332Gを含む正常配列,14クローンが17塩基を欠失した配列,2クローンが48塩基を欠失した配列であり(Fig. 1B),c.1332G>Aはスプライシング変異であることが確認された.17塩基欠失はframeshiftを起こすことから,生成タンパクは失活すると考えられる.48塩基欠失はin frameであるが,得られた発現クローン数が少ないことから,mRNA品質管理機構による排除が示唆された.さらに,c.1332Gを含む正常配列ではc.1349G>A変異を伴っていた一方,17塩基を欠失した配列と48塩基を欠失した配列ではc.1349G配列であった.患者両親の遺伝子解析は実施していないが,酵素活性測定結果と合わせ,本症例はc.1349G>A (R450H)およびc.1332G>Aの複合へテロ接合によるVLCAD欠損症と結論した.
患者はその後,安静と頻回の炭水化物摂取により,横紋筋融解症の再燃はなく経過した.妊娠37週2日時点で,硬膜外麻酔併用での吸引分娩により2,940 gの健常男児を出産した.児には神経学的な異常所見はなく,児のタンデムマススクリーニングには異常なかった.患者は出産後にカルニチン1,500 mg/日の内服補充,飢餓や過剰な運動を避ける,炭水化物の頻回摂取などの生活指導を行っており,育児で多忙となる時や睡眠不足の際には,血清CK 1,000 IU/l程度に上昇することもありながら,現在まで腎機能障害を起こすことなく,安定して経過している.
考察
VLCAD欠損症は脂肪酸代謝異常症の一つであり,空腹時や発熱,運動,下痢・嘔吐などの脂肪酸代謝に負荷がかかる状態が生じると,急性代謝不全を引き起こしやすく,心筋や骨格筋,肝臓などに臓器障害が生じる.VLCAD欠損症は,アシルカルニチン分析でのC14:1の上昇,C14:1/C2比の上昇,ACADVL遺伝子変異,末梢血リンパ球や培養皮膚線維芽細胞培養液中のアシルカルニチン分析やpalmitoyl-CoA dehydrogenase活性評価を行うことで診断される4)10).新生児期発症型や乳幼児期発症型のVLCAD欠損症は,飢餓により低血糖や肝機能障害で発症することが多い5)が,遅発型では,学童期以降に骨格筋への負荷を誘因とした横紋筋融解症やミオパチーなどで発症することが多い7).成人期の軽症例では,発作間欠期の血清アシルカルニチン分析で異常所見を認めないことがあるため,発作期の血清アシルカルニチン分析に加えて,遺伝子検査や末梢血リンパ球,培養皮膚線維芽細胞などを用いた脂肪酸代謝能の評価が重要であり,これらの検査のうち一つが陽性であれば確定診断となる4).本症例では,CK値が上昇している時点でアシルカルニチン分析を行い,C14:1とC14/C2比の上昇を認めたため,VLCAD欠損症を疑うことができた.また,本症例は,父親に運動後褐色尿の病歴があるものの,VLCAD欠損症は常染色体潜性遺伝形式をとるため,父親の病歴が直接関係あるかはわからなかった.
本症例は,末梢血リンパ球のpalmitoyl-CoA dehydrogenase活性値が正常者平均の6.5%と低値であった.Palmitoyl-CoA dehydrogenase活性はVLCAD欠損症で低値を示し,診断に有用であることが示されているが10),一般に,酵素活性が低いほど病状が悪化する傾向があるとされる11).VLCAD欠損症では,遺伝子変異の種類によって酵素の残存活性が変化することが知られているが8),本症例で認められたc.1332G>A変異は,ホモ変異の場合には高度に酵素活性が低下し,致死的な経過をとる9).VLCAD欠損症では,遺伝子変異のタイプとその組み合わせによって,酵素の残存活性が様々に変わることが知られている8).c.1332G>A変異は同義置換ではあるものの,イントロンとエクソンの接合部に位置しており,スプライス異常を惹起するため酵素活性が高度に低下するものと考えられた.我々の症例の場合は,主としてp.R450H変異酵素に由来すると推定される残存活性によって,遅発骨格筋型の臨床像を呈したと考えられた.
Table 1
Acylcarnitine concentration on tandem mass spectrometry.
| Acylcarnitine |
Concentration
(nmol/ml) |
Control
(nmol/ml) |
| C0 |
7.26 |
>10 |
| C2 |
3.32 |
— |
| C3 |
0.52 |
<3.5 |
| C4 |
0.04 |
<1 |
| C5 |
0.02 |
<1 |
| C6 |
0.02 |
<0.3 |
| C8 |
0.02 |
<0.3 |
| C10 |
0.04 |
<0.35 |
| C14 |
0.26 |
<0.40 |
| C14:1 |
0.84 |
<0.40 |
| C16 |
0.54 |
<6.0 |
| C16-OH |
0 |
<0.05 |
| C18 |
0.27 |
<3.0 |
| C18:1 |
0.54 |
<3.0 |
| C18:1-OH |
0.01 |
<0.05 |
また,VLCAD欠損症に関する妊娠・出産の症例報告は,文献検索した範囲では6例が報告されている(Table 2)12)~17).全例が初産で,骨格筋症状の悪化や再燃を伴った.多くの症例は出産前に骨格筋症状,反復性のCK上昇などからVLCAD欠損症と診断されていたが,Murataらは本症例と同様,初産婦のCK上昇を契機にVLCAD欠損症と診断された症例を報告している13).このように,未診断のVLCAD欠損症が妊娠悪阻を契機に悪化する可能性があり,注意が必要である.治療としては,ブドウ糖液による点滴治療や炭水化物の頻回摂取などが行われ,半数は経腟分娩,半数は帝王切開で安全に出産できていた.栄養療法として,VLCAD欠損症の増悪を避けるため,炭水化物や中鎖脂肪酸の頻回摂取が有用とされており18),本症例でも妊娠中に頻回の炭水化物摂取を奨励し,骨格筋症状の再燃を予防した.
本邦では,先天性代謝性疾患の検索のため新生児マススクリーニングが行われており,2014年からはタンデムマススクリーニング法が導入され5),VLCAD欠損症を含む脂肪酸代謝異常症が発症前に発見されるようになっている.ただし,本疾患はマススクリーニングでは全例を発見することはできないとされ5),未診断のまま成人となる症例が存在しうるため,横紋筋融解症を繰り返す症例では,脂肪酸代謝異常症をはじめとする先天性疾患も広く鑑別に挙げる必要がある.
Table 2
Previous reports of very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency associated with pregnancy.
| First author |
Year |
Age (years) |
Gravidity |
Age at diagnosis (years) |
Symptoms |
CK peak (IU/ml) |
Treatment |
Delivery style |
| Mendez-Figueroa H12) |
2010 |
21 |
G1P0 |
20 |
myalgia and muscle weakness |
437 |
intravenous glucose infusion |
vaginal delivery |
| Murata KY13) |
2014 |
34 |
primigravida |
undiagnosed |
heart failure |
21,530 |
treatment for heart failure |
cesarean section |
| Yamamoto H14) |
2015 |
17 |
not mentioned |
12 |
myalgia |
3,388 |
intravenous glucose infusion |
vaginal delivery |
| Yamada K15) |
2019 |
31 |
not mentioned |
26 |
fatigue |
3,934 |
frequent juice intake |
cesarean section |
| Endo E16) |
2021 |
27 |
G1P0 |
21 |
asymptomatic |
274 |
intravenous glucose infusion |
vaginal delivery |
| Akar HT17) |
2021 |
32 |
not mentioned |
23 |
asymptomatic |
29,703 |
intravenous glucose infusion |
cesarean section |
| Our case |
2023 |
25 |
G1P0 |
undiagnosed |
myalgia and muscle weakness |
11,755 |
frequent carbohydrate intake |
vaginal delivery |
Abbreviations: CK, creatine kinase; G, gravida; P, pregnancy.
結語
妊娠悪阻を契機に再燃し,診断に至ったVLCAD欠損症の1例を経験した.遺伝子診断では,複合ヘテロ変異を認め,特にc.1332G>A変異はアミノ酸配列変化を伴わないサイレント変異と当初考えたが,変異部位がイントロンとエクソンの接合部に位置しており,mRNA分解を介した疾患への関与が考えられた.臨床的,分子病態学的に重要な症例と考えられた.
Acknowledgments
謝辞:本症例の診断,治療に関しましてご指導頂きました九州大学名誉教授 吉良潤一先生に深謝いたします.
Notes
※著者全員に本論文に関連し,開示すべきCOI状態にある企業,組織,団体はいずれも有りません.
文献
- 1) Knottnerus SJG, Bleeker JC, Wüst RCI, et al. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev Endocr Metab Disord 2018;19:93-106.
- 2) Aoyama T, Uchida Y, Kelley RI, et al. A novel disease with deficiency of mitochondrial very-long-chain acyl-CoA dehydrogenase. Biochem Biophys Res Commun 1993;191:1369-1372.
- 3) Laforet P, Acquaviva-Bourdain C, Rigal O, et al. Diagnostic assessment and long-term follow-up of 13 patients with very long-chain acyl-coenzyme A dehydrogenase (VLCAD) deficiency. Neuromuscul Disord 2009;19:324-329.
- 4) 小林弘典.【新ガイドラインの理解を深める 新生児マススクリーニング】VLCAD欠損症.小児科診療 2021;84:267-272.
- 5) 日本先天代謝異常学会.新生児マススクリーニング対象疾患等診療ガイドライン2015.東京:診断と治療社;2015.
- 6) Shibata N, Hasegawa Y, Yamada K, et al. Diversity in the incidence and spectrum of organic acidemias, fatty acid oxidation disorders, and amino acid disorders in Asian countries: Selective screening vs. expanded newborn screening. Mol Genet Metab Rep 2018;16:5-10.
- 7) Fukao T, Watanabe H, Orii K, et al. Myopathic form of very-long chain acyl-coa dehydrogenase deficiency: evidence for temperature-sensitive mild mutations in both mutant alleles in a Japanese girl. Pediatr Res 2001;49:227-231.
- 8) Osawa Y, Kobayashi H, Tajima G, et al. The frequencies of very long-chain acyl-CoA dehydrogenase deficiency genetic variants in Japan have changed since the implementation of expanded newborn screening. Mol Genet Metab 2022;136:74-79.
- 9) Yamamoto A, Nakamura K, Matsumoto S, et al. VLCAD deficiency in a patient who recovered from ventricular fibrillation, but died suddenly of a respiratory syncytial virus infection. Pediatr Int 2013;55:775-778.
- 10) Tajima G, Sakura N, Shirao K, et al. Development of a new enzymatic diagnosis method for very-long-chain Acyl-CoA dehydrogenase deficiency by detecting 2-hexadecenoyl-CoA production and its application in tandem mass spectrometry-based selective screening and newborn screening in Japan. Pediatr Res 2008;64:667-672.
- 11) Hesse J, Braun C, Behringer S, et al. The diagnostic challenge in very-long chain acyl-CoA dehydrogenase deficiency (VLCADD). J Inherit Metab Dis 2018;41:1169-1178.
- 12) Mendez-Figueroa H, Shchelochkov OA, Shaibani A, et al. Clinical and biochemical improvement of very long-chain acyl-CoA dehydrogenase deficiency in pregnancy. J Perinatol 2010;30:558-562.
- 13) Murata KY, Sugie H, Nishino I, et al. A primigravida with very-long-chain acyl-CoA dehydrogenase deficiency. Muscle Nerve 2014;49:295-296.
- 14) Yamamoto H, Tachibana D, Tajima G, et al. Successful management of pregnancy with very-long-chain acyl-coenzyme A dehydrogenase deficiency. J Obstet Gynaecol Res 2015;41:1126-1128.
- 15) Yamada K, Matsubara K, Matsubara Y, et al. Clinical course in a patient with myopathic VLCAD deficiency during pregnancy with an affected baby. JIMD Rep 2019;49:17-20.
- 16) 遠藤英作,小畠真奈,宮代夢子ら.極長鎖アシルCoA脱水素酵素欠損症(骨格筋型)合併妊娠の一例.日本周産期・新生児医学会雑誌 2021;57:205-208.
- 17) Akar HT, Cagan M, Yildiz Y, et al. Complicated peripartum course in a patient with very long-chain acyl-coenzyme A dehydrogenase (VLCAD) deficiency. Neuromuscul Disord 2021;31:566-569.
- 18) 春木明代,川井元晴,小笠原淳一ら.極長鎖アシルCoA脱水素酵素(VLCAD)欠損症の22歳女性例に対する食事療法の試み.臨床神経 2010;50:172-174.
