要旨
症例は生来健康な44歳男性.両親にいとこ婚あり.42歳頃から両足指の脱力が出現し,同時期から両足部のチアノーゼを自覚.その後,症状進行し44歳時には走れなくなった.両足部に顕著なチアノーゼと下肢遠位筋脱力とアキレス腱反射の消失を認め,神経伝導検査で軸索型多発ニューロパチーを認めた.網羅的遺伝子検査によりmembrane metalloendopeptidase(MME)遺伝子変異を伴うautosomal-recessive Charcot–Marie–Tooth disease 2Tと診断した.本例の足部チアノーゼは高度であり背景病態にMME遺伝子変異との関連が推測された.
Abstract
The patient is a 44-year-old man. His parents are consanguineous. He experienced muscle weakness in his toe and distal tingling sensation in his feet at 42 years of age, which gradually progressed. Additionally, a marked cyanotic discoloration of the feet appeared and worsened progressively. Neurological examination revealed loss of tendon reflexes and distal muscle weakness in the lower extremities. Findings from nerve conduction studies indicated axonal polyneuropathy. Upon detection of the MME gene mutation, the patient was diagnosed with autosomal-recessive Charcot–Marie–Tooth disease 2T (ARCMT2T). In this case, cyanosis of the lower extremities possibly was associated with ARCMT2T, and it was suggested to be due to neprilysin deletion linked with the MME mutation. This represents the first documented occurrence of cyanosis as a distinctive feature of CMT with MME mutation.
はじめに
現在までに複数のCharcot–Marrie–Tooth病(Charcot–Marrie–Tooth disease,以下CMTと略記)の原因遺伝子が特定され1),CMTの病態解明の端緒となり新規治療標的としての可能性が示されてきた.Higuchiらによって明らかにされたmembrane metalloendopeptidase(MME)遺伝子変異は本邦で報告されている成人発症常染色体劣性遺伝型CMTのなかで最も頻度の高い変異であり,Automosal Reccesive Charcot–Marrie–Tooth 2T(AR-CMT2T)が提唱されている1).われわれは中年以降に発症し顕著な足部チアノーゼを呈したMME遺伝子変異を伴うCMT症例を経験した.チアノーゼがAR-CMT2Tの臨床診断上重要な所見であると考えられ文献的考察を加えて報告する.
症例
患者:44歳,男性
主訴:両足の脱力と異常感覚,両足のチアノーゼ
既往歴:腰椎椎間板ヘルニア.
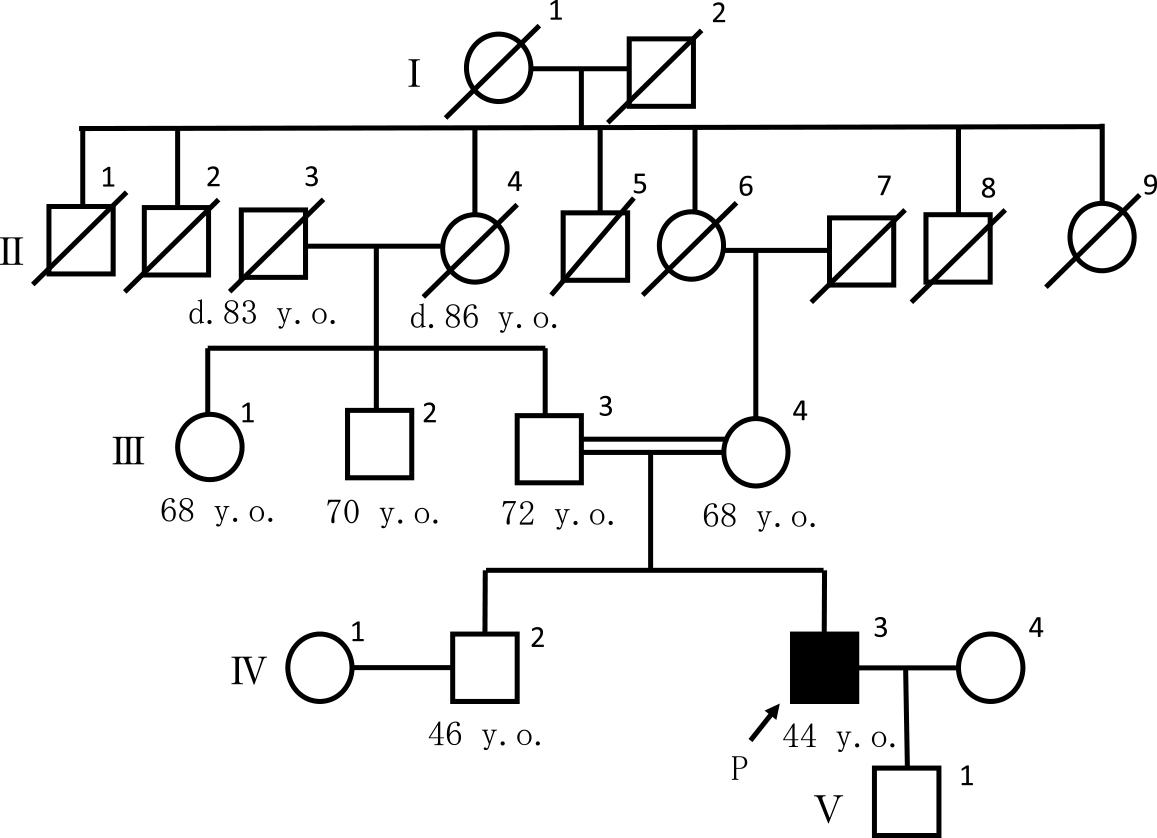
家族歴:両親がいとこ婚(Fig. 1).血縁者に類症なし.
現病歴:出生・発育に問題はなかった.42歳頃から両足の第1足趾を背屈させられなくなり,歩行時に躓きやすくなった.その後も両足部の脱力とジンジン感を自覚する様になり歩きづらくなっていた.また同時期から両足部のチアノーゼを自覚していた.両足部の脱力は徐々に増悪し,44歳時には小走りすることが出来なくなった.
入院時現症:一般身体所見:身長177.3 cm,体重79.9 kg,BMI 25.4,血圧134/84 mmHg,脈拍74 bpm整,呼吸回数16回/分,体温36.4°C.両足部冷感とチアノーゼを認めた(Fig. 2A).大腿動脈,膝窩動脈,両足背動脈の触知は可能であった.凹足などの足部変形はなし.
神経学的所見:意識清明で認知機能は正常であり,脳神経系に異常所見を認めなかった.握力は右手37.5 kg/左手37.8 kgで両上肢の筋力は正常であった.徒手筋力テストでは腸腰筋5/5,大腿四頭筋5/5,大腿屈筋5/4,前脛骨筋4/4,腓腹筋4+/4,足趾伸筋2/2,足趾屈筋2/2と両下肢遠位部優位に筋力低下を認めた.両側の前脛骨筋,腓腹筋に軽度の筋萎縮を認めた.歩容は正常だが踵歩きと爪先立ちは出来なかった.腱反射についてはアキレス腱反射が両側で消失していた.病的反射は陰性であった.感覚系では足関節以遠に軽度の異常感覚と深部覚低下が認められた.触覚と温痛覚は正常であった.自律神経系に関しては性機能,発汗,排便排尿は正常で起立性低血圧を認めなかった.
検査所見:血算,生化学,内分泌検査,凝固は正常で,抗核抗体など膠原病関連の自己抗体及びM蛋白は認めず,血清VEGFも正常値であった.抗ガングリオシド抗体は抗GM2 IgG抗体と抗GalNac-GD1a IgG抗体が陽性(++)で,抗GalNac-GD1a IgM抗体が弱陽性(+)であった.脳脊髄液検査では細胞数2/μl(単形核),蛋白46.1 mg/dl,IgG index 0.50,オリゴクローナルバンド陰性であった.神経伝導検査では上肢の感覚神経は正常で運動神経伝導速度が軽度低下していた(Table 1).下肢神経の複合筋活動電位(compound muscle action potential,以下CMAPと略記)は著明に低下しており,脛骨神経に関しては近位刺激では誘発することが出来なかった(Table 1).腓骨神経で伝導速度の低下を伴っていたが伝導ブロックや時間的分散は認めなかった.針筋電図検査では第一背側骨間筋,前脛骨筋において線維束自発電位を認め,第一背側骨間筋には線維自発電位と陽性鋭波を認めた.随意収縮時は遅延動員と干渉不良を認め神経原性変化が考えられた.以上より,軸索型ニューロパチーを示唆する所見であると考えられた.腰椎MRIでは神経根の腫大や造影効果を認めなかった.全身CTでは臓器腫大や骨硬化像などの異常所見は認められなかった.両足部のチアノーゼを伴っていたため末梢血管系の異常も考慮し血圧脈波検査(Ankle branch index: ABI),下肢動脈エコー検査などを施行したが末梢血管障害を示唆する所見を認めなかった.
Table 1
Results of nerve conduction study (peak to peak).
| Nerve |
Side |
Motor nerve |
|
Sensory nerve |
DL
(msec) |
CMAP (mV)
Distal |
CMAP (mV)
Proximal |
MCV
(m/sec) |
F-latency
(msec) |
F-freq
(%) |
SNAP
(μV) |
SCV
(m/sec) |
| Median |
R |
4.0 |
8.4 |
8.4 |
47.2 |
34.6 |
37 |
|
27.1 |
51.1 |
| Ulnar |
R |
3.2 |
9.1 |
9.2 |
44.6 |
36.8 |
81 |
25.2 |
51.2 |
| Tibial |
R |
5.9 |
0.061 |
N.E. |
N.E. |
N.E. |
0 |
|
|
| Peroneal |
R |
6.7 |
0.32 |
0.33 |
21.9 |
|
|
|
|
| Sural |
R |
|
|
|
|
|
|
4.70 |
36.6 |
DL: distal latency, CMAP: compound muscle action potential, MCV: motor nerve conduction velocity, F-freq: F wave frequency, SNAP: sensory nerve action potential, SCV: sensory nerve conduction velocity, N.E.: not evoked, R: right.
臨床経過:本例はいとこ婚の家族歴が有り,緩徐進行性の臨床経過,両下肢遠位部に対称性の運動感覚障害と軽度の筋萎縮を呈していた.両下肢では神経伝導検査での軸索障害が著明でありCMTが疑われた.鹿児島大学での次世代シークエンサーを用いたエクソーム解析によりCMTの網羅的遺伝子解析を行いMME遺伝子変異(c.655-2A>G, homozygouosの変異)に伴うCMTと診断した(本症のバリアントは既報告の変異であった.また遺伝子検査は本人の承諾を得て施行した).
考察
MME遺伝子変異は2016年にHiguchiらにより報告されたCMTの原因遺伝子である2).2022年の報告では2021年5月までに鹿児島大学で集積されたPMP22重複/欠失を除く798例のCMT患者の遺伝子変異のうち28例でMME遺伝子変異が認められており,常染色体劣性遺伝のCMTのなかでは最も頻度の高い変異であった.さらにHiguchiらはMME遺伝子変異に伴うCMT患者10名の臨床的特徴についても報告しており,発症時期は遅発性(発症年齢36~56歳,中央値50歳)で,10名のうち6名の両親にはいとこ婚が認められた.臨床症状に関しては全例で緩徐進行性の四肢遠位筋の脱力と同部位の筋萎縮,歩行障害,四肢遠位部の感覚障害,腱反射低下または消失が認められ,錐体路徴候や小脳失調,認知機能障害などの中枢神経症状を呈した症例はなく典型的なCMTの特徴と合致していた.神経伝導検査では1例で正中神経伝導速度が軽度遅延(37.4 m/s)を呈したため脱髄型もしくは中間型CMTと診断されたが,その他の9例は軸索型CMTの所見であった(正中神経運動神経の平均値潜時4.12 s[3.2~6.3 s],distal CMAP 5.34 mV[1.7~10 mV],MCV 43.8 m/s[37.4~53 m/s]であった).神経生検の病理所見についてはonion bulb形成は認められず有髄線維密度の著明な減少が認められ軸索型ニューロパチーを支持する所見であった.本例の神経伝導検所見は下肢でCMAP及び複合感覚神経電位の高度低下を呈しており,上肢では僅かなMCVの低下を伴っていたが軸索型CMTの範疇として矛盾しないレベルであり,臨床像と合わせて典型的なMME遺伝子変異によるCMTであると考えられた(Table 2).
Table 2
Genetic, clinical, and laboratory findings.
|
Our case |
Report example (n = 10) |
| Age, year |
44 |
Median 50 (36–56) |
| Sex |
Men |
Men (80%), Female (20%) |
| Inheritance pattern |
AR |
AR |
| Consanguinity |
+ |
+ (60%) |
| Initial symptom |
Motor |
Motor 80%, Sensory 20% |
| MMT (distal lower limbs) at first visit |
4 |
1–4 |
| Sensory disturbance |
+ |
+ (90%) |
| Decreased DTRs |
+ |
+ (100%) |
| Cyanosis |
+ |
No data |
| Dementia |
− |
− |
| Brain atrophy |
− |
− |
| Type |
Axonal neuropathy |
Axonal neuropathy (90%) demyelinating
and axonal neuropathy (10%) |
AR = autosomal reccesive; DTRs = deep tendon reflexes.
MME遺伝子異常による末梢神経障害の機序については十分に明らかにされていない.MME遺伝子はneprilysin(NEP)をコードしており,このNEPはヒトを含む哺乳類の中枢神経系と末梢神経系で多く発現していることが明らかにされている2)3).中枢神経系ではアミロイドβの分解酵素として作用することから,近年NEPの機能低下がAlzheimer病発症に関連していることが報告されている4).一方で末梢神経系においてNEPは軸索・髄鞘の両方に発現しているが,髄鞘により多く発現していることが明らかにされており,健常者ではNEPが末梢神経の発生に関与することや軸索損傷時にNEPが末梢神経で増加し軸索再生に関連することが推測されている1)5).このことからMME遺伝子変異を伴うCMTにおいては末梢神経の発生に何らかの異常が生じていることや軸索再生が正常に行われず軸索障害が進行していると推測される.
本例の特徴として著明な足部のチアノーゼを認めたが,Higuchiらの報告例にはチアノーゼの記載はないためAR-CMT2Tにおけるチアノーゼの頻度や程度については不明である.
CMTでは足部のチアノーゼが出現することは知られているが6),CMTにおけるチアノーゼに関するまとまった報告はなくCMTの病型や遺伝子異常とチアノーゼの合併頻度や程度との関連などは不明である.本例のチアノーゼの特徴としては病初期から出現し,チアノーゼの程度が運動感覚障害の程度と相関せず顕著であった点である.Fig. 2BにはCMT type 1A(自験例)で認められたチアノーゼとの比較を提示している(Fig. 2B).このようなチアノーゼの合併がMME遺伝子異常と関連するかは不明であるが,NEPがエンケファリン,サブスタンスP,心房性ナトリウム利尿ペプチドなどの多くの基質の分解に関与していることから,MME遺伝子異常が血管機能の異常を引き起こす可能性がある7).MMEノックアウトマウスでの実験ではNEP欠損に伴いその基質であるエンドセリン1やサブスタンスPといった血管作動性を有する物質の産生が毛細血管で上昇することが報告されている8).特にエンドセリン1は末梢血管収縮を,サブスタンスPは血管透過性亢進を惹起することから,こうした物質の過剰がcold complex regional pain syndrome(CRPS)様のチアノーゼ,冷感,浮腫などの症状を引き起こすと考えられている8).本例のチアノーゼの病態としてもMME遺伝子異常を背景としてNEPの機能異常による末梢循環障害を生じている可能性が推測された.一方で本例は軸索型CMTであることから小径無髄線維である自律神経の障害が下肢遠位部で生じていた可能性はある.しかしながら,MME遺伝子異常を伴うCMTにおける皮膚生検の解析では小径無髄線維の異常が指摘されていない9).本症例では臨床的に自律神経障害を認めていないものの,精密な自律神経系の解析を行っていないため足部のチアノーゼにおける自律神経障害の関与については不明である.
本例では抗ガングリオシド抗体が陽性であったが,抗GM2 IgG抗体についてはcytomegalovirus感染後の顔面神経麻痺を伴う脱髄型のギラン・バレー症候群との関連性が,抗GalNAc-GDla IgG抗体についてはacute motor axonal neuropathyとの関連性が報告されているが,両者とも慢性進行性軸索型ニューロパチーでの報告はなく本例におけるこの抗体の意義は不明である10).一方でCMT患者では慢性炎症性脱髄性多発神経炎が合併しやすく11),免疫療法が部分的に有効であることが報告されている12).抗ガングリオシド抗体が陽性であったことから本例でも同様の背景病態を伴っている可能性を考慮してγグロブリン大量静注療法を施行したが臨床症状と神経伝導検査では変化を認めなかった.以上からは本例における抗ガングリオシド抗体の臨床的意義は乏しいと考えられる.MME遺伝子変異を有するCMTではMME遺伝子から産生されるNEPが髄鞘にも多く発現していることから髄鞘にも脆弱性が存在することが推測され,長期間にわたり髄鞘破壊による髄鞘成分が比較的多く免疫系に曝露されたことで抗ガングリオシド抗体が二次的に産生された可能性はある.
結語
本例で認められた顕著なチアノーゼはMME遺伝子変異を伴うCMT症例ではこれまで報告されていないが,MME遺伝子変異を伴うCMTの重要な所見である可能性があり,今後の症例の集積が必要であると考えられる.
Acknowledgments
謝辞:ガングリオシド抗体を測定いただきました近畿大学脳神経内科楠進先生および本例におけるガングリオシド抗体に関する貴重なご助言をいただきました山口大学脳神経内科古賀道明先生に深謝いたします.
Notes
本報告の要旨は,第237回日本神経学会九州地方会で発表し,会長推薦演題に選ばれた.
※著者全員に本論文に関連し,開示すべきCOI状態にある企業,組織,団体はいずれも有りません.
文献
- 1) Higuchi Y, Takashima H. Clinical genetics of Charcot–Marie–Tooth disease. J Hum Genet 2023;68:199-214.
- 2) Higuchi Y, Hashiguchi A, Yuan J, et al. Mutations in MME cause an autosomal-recessive Charcot-Marie-Tooth disease type 2. Ann Neurol 2016;79:659-672.
- 3) Kioussi C, Crine P, Matsas R, et al. Endopeptidase-24.11 is suppressed in myelin forming but not in non-myelin-forming Schwann cells during development of the rat sciatic nerve. Neuroscience 1992;50:69-83.
- 4) Fujisawa M, Sano Y, Kanda T, et al. Charcot-Marie-Tooth disease type 2 caused by homozygous MME gene mutation superimposed by chronic inflammatory demyelinating polyneuropathy. Clin Neurol 2017;57:515-520.
- 5) Kioussi C, Mamalaki A, Matsas R, et al. Expression of endopeptidase-24.11(common acute lymphoblastic leukaemia antigen CD10) in the sciatic nerve of the adult rat after lesion and during regeneration. Eur J Neurosci 1995;7:951-961.
- 6) Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol 2009;8:654-667.
- 7) Yamamoto K, Rakugi H. Angiotensin receptor-neprilysin inhibitors: comprehensive review and implications in hypertension treatment. Hypertens Res 2021;44:1239-1250.
- 8) Heidrun H, Lan H, Sommer C, et al. Increased pain and neurogenic inflammation in mice deficient of neutral endopeptidase. Neurobi Dis 2009;35:177-183.
- 9) Grumbach M, Togel S, Schabhuttl M, et al. Rare variants in MME, Encording metalloprotease neprilysin, are linked to late-onset autosomal-dominant axonal polyneuropathies. Am J Hum Genet 2016;99:607-623.
- 10) Kaida K, Kusunoki S, Kamakura K, et al. GuillainBarré syndrome with antibody to a ganglioside. N-acetylgalactosaminyl GDla. Brain 2000;123:116-124.
- 11) Ginsberg L, Malik O, Kenton A, et al. Coexistent hereditary and inflammatory neuropathy. Brain 2004;127:193-202.
- 12) Yasuo M, Masahiko T, Masayuki B, et al. A family with IVIg-responsive Charcot-Marie-Tooth disease. J Neurol 2013;260,1147-1151.