要旨
末梢神経障害を合併したBAG3変異による筋原線維性ミオパチー(myofibrillar myopathy,以下MFMと略記)の1例を経験したので報告する.症例は19歳の女性で,階段昇降や歩行障害が進行した.神経学的に体幹筋と両下肢遠位筋優位の筋力低下を認め,筋生検で縁取り空胞や不整な筋線維配列を確認した.遺伝子解析でBcl2-Associated Athanogene 3(BAG3)の病的ヘテロ接合性変異(p.P209L)が判明し,MFM6と診断した.本症は小児期に発症し心筋症を合併する予後不良の稀なミオパチーであるが,軸索型ニューロパチーを合併し,尖足拘縮,体幹屈筋が弱い特徴がある.若年発症のニューロミオパチーでは本疾患を疑い,遺伝子検査を検討すべきである.
Abstract
A 19-year-old female, normal at birth, grew up without neck movement when getting up. She needed a handrail to climb stairs since the age of 10 years old, and walked slowly since the age of 16 years old. Neurological examination revealed loss of deep tendon reflexes, decreased vibratory sensation, weakness of distal muscles of the lower extremities, and weakness of mainly cervical trunk muscles suspected to be due to myopathy. Nerve conduction studies suggested axonal polyneuropathy, and needle EMG showed short duration MUP, myotonic discharge, and rimmed vacuoles on muscle biopsy. Genetic analysis revealed a previously reported pathological mutation (p.P209L, heterozygous) in Bcl2-Associated Athanogene 3 (BAG3), and a diagnosis of MFM6 was made. P209L is a poor prognosis myopathy that develops in childhood and is associated with cardiomyopathy. P209L is a solitary myopathy associated with axonal neuropathy and characterized by apex foot contracture and weak neck to trunk flexion. This disease is suspected in young-onset neuromyopathy.
はじめに
筋原線維性ミオパチー(myofibrillar myopathy,以下MFMと略記)は,筋原線維の不整を特徴とする遺伝性ミオパチーである.筋組織学的な特徴は,Z帯の崩壊,次いで筋原線維の破壊と蛋白質の異所性蓄積である1).Bcl2-Associated Athanogene 3(BAG3)遺伝子の変異は,進行性の近位および体幹のミオパチー,心筋症,呼吸障害を特徴とするMFM6型(MFM6)を引き起こす.一方で,遺伝性運動感覚ニューロパチーであるCharcot–Marie–Tooth病(CMT)を引き起こすことも報告されている2).MFM6は本邦での報告は少ない.そこで今回我々は,BAG3遺伝子にc.626C>T (p.Pro209Leu)変異を有し,近位筋や体幹の筋力低下に加え,軸索障害を主体とした運動感覚性ニューロパチーの臨床像を呈したMFM6の症例を報告する.
症例
症例:19歳 女性
主訴:歩きにくい,階段が上れない
既往歴:なし.
家族歴:近親者に同症状はない.母に低身長があるが,神経診察上,低身長以外に患者と同様の徴候を認めない.
現病歴:周産期異常や精神発達遅滞はなかったが,乳児期より首がすわらず,現在に至るまで起居動作時に首がついてこないため振りかぶるようにして頭部を上げていた.それ以外には運動発達に大きな異常はなかった.10歳頃には走るのが遅くなり,階段を上るのに手すりを使うようになった.16歳頃には歩行が遅いことを自覚した.19歳ではアルバイト先で物を運ぶ仕事に時間がかかることに気がつき,階段昇降もできなくなっていた.近医受診の際にCK 1,280 U/lと高値を認め,筋疾患が疑われたため,同年当院受診し精査入院となった.
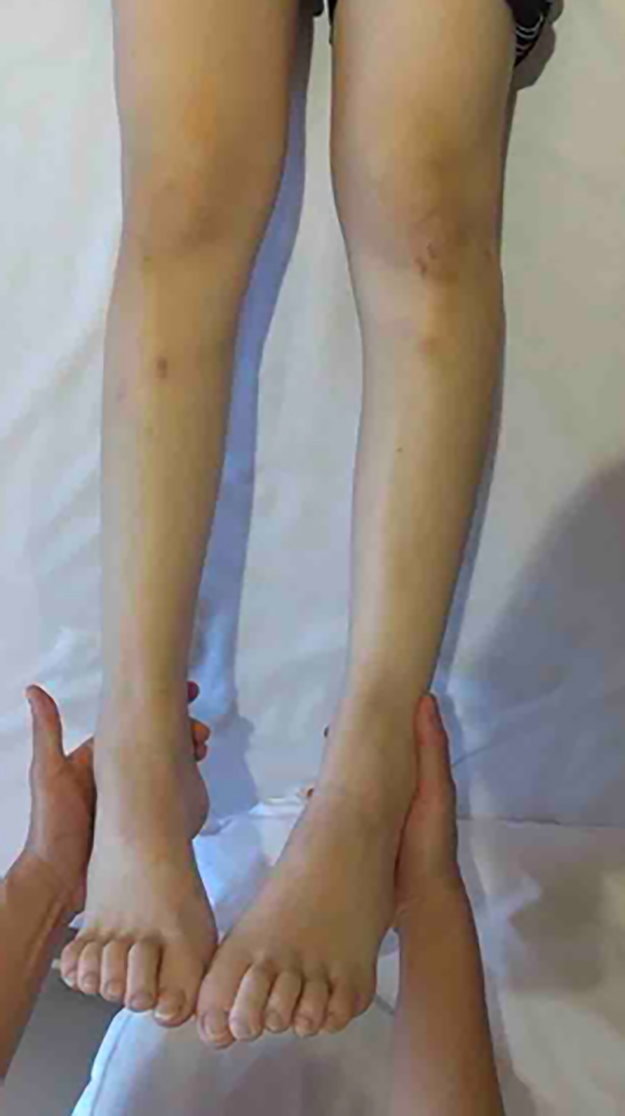
一般身体所見:身長142.3 cm,体重37.9 kg,BMI 18.7と低身長および低体重を認めた.左翼状肩甲,尖足,両側足趾の槌状変形と凹足を認めた(Fig. 1).
神経学的所見:脳神経系では正中視で両側内斜視を呈し,眼球運動は両眼の軽度外転障害と,左眼に左注視方向性眼振を認めた.顔面筋力は正常で,嚥下・構音機能も正常であった.徒手筋力検査(manual muscle testing,以下MMTと略記)では,頸屈4,腹直筋2と体幹筋の筋力低下を認め,仰臥位から腹筋運動の際,背中を浮かすことができない状態であった.また,三角筋,腸腰筋,中臀筋,大腿二頭筋などの近位筋にMMT 4の筋力低下を認め,Gowers徴候が陽性であった.前脛骨筋MMT 2,腓腹筋MMT 4と下肢遠位筋が低下しており,筋萎縮は両大腿と下腿に認めた.深部腱反射はすべて消失していた.触覚や温痛覚,位置覚の低下はなかったが,振動覚は上肢,下肢(外果)ともに7秒に低下していた.起立は柵を把持しながら勢いをつけて行うが,立位保持に問題なく,Romberg試験も陰性だった.歩行は鶏歩,動揺性歩行を認めた.
検査所見:末梢血には異常がなく,生化学ではCK 754 U/l(基準値41~153),CK-MB 64 U/l(基準値 <12),ミオグロビン87.7 ng/ml(基準値 <70),アルドラーゼ10.3 U/l(基準値2.7~7.5)と高値を認めた.CRP 0.02 mg/dl,HbA1c 5.3%,血糖値は86 mg/dlと正常だった.膠原病や代謝性疾患のスクリーニング検査は陰性だった.血液ガス分析ではPaO2 94.3 mmHg,PaCO2 40.2 mmHgと正常であった.心電図,経胸壁心エコー検査,胸部レントゲンで異常は認められなかった.呼吸機能検査では,肺活量(VC)が1.82 l(60.1%),1秒率(FEV 1.0%(G))96.8%と拘束性障害を認め,呼吸筋障害が示唆された.
電気生理学的検査:神経伝導検査では運動神経にて右正中神経の伝導遅延(DL = 5.9 ms,MCV = 45.5 m/s)および右尺骨神経の振幅低下(CMAP = 5.0 mV)と脛骨神経の振幅低下(CMAP = 0.3 mV)を認め,感覚神経は全て導出できなかった(Table 1).振幅低下が主体の所見であり,軸索障害が示唆された.針筋電図検査は,近位筋(上腕二頭筋,外側広筋)と前脛骨筋にて行い,安静時の線維自発電位とミオトニー放電を認めた.運動単位電位(MUP)はunstable polyphasic patternがめだつshort duration MUPであり,活動性の筋原性変化が示唆され,神経原性変化は認めなかった.
Table 1
Nerve conduction study.
| Nerve |
Motor nerve |
Sensory nerve |
| DL (ms) |
MCV (m/s) |
dCMAP (mV) |
minimal F-wave latency (ms) |
SNAP |
| Rt Median N |
5.9 |
45.5 |
5.4 |
26.4 |
N.D. |
| Rt Ulnar N |
3.6 |
49.7 |
5.0 |
29.4 |
N.D. |
| Rt Tibial N |
5.7 |
37.2 |
0.3 |
|
|
| Rt Sural N |
|
|
|
|
N.D. |
Rt: right, N: nerve, DL: Distal latency, MCV: motor nerve conduction velocity, dCMAP: distal compound muscle action potential (peak to base), ND: not detected, SNAP: sensory nerve action potential.
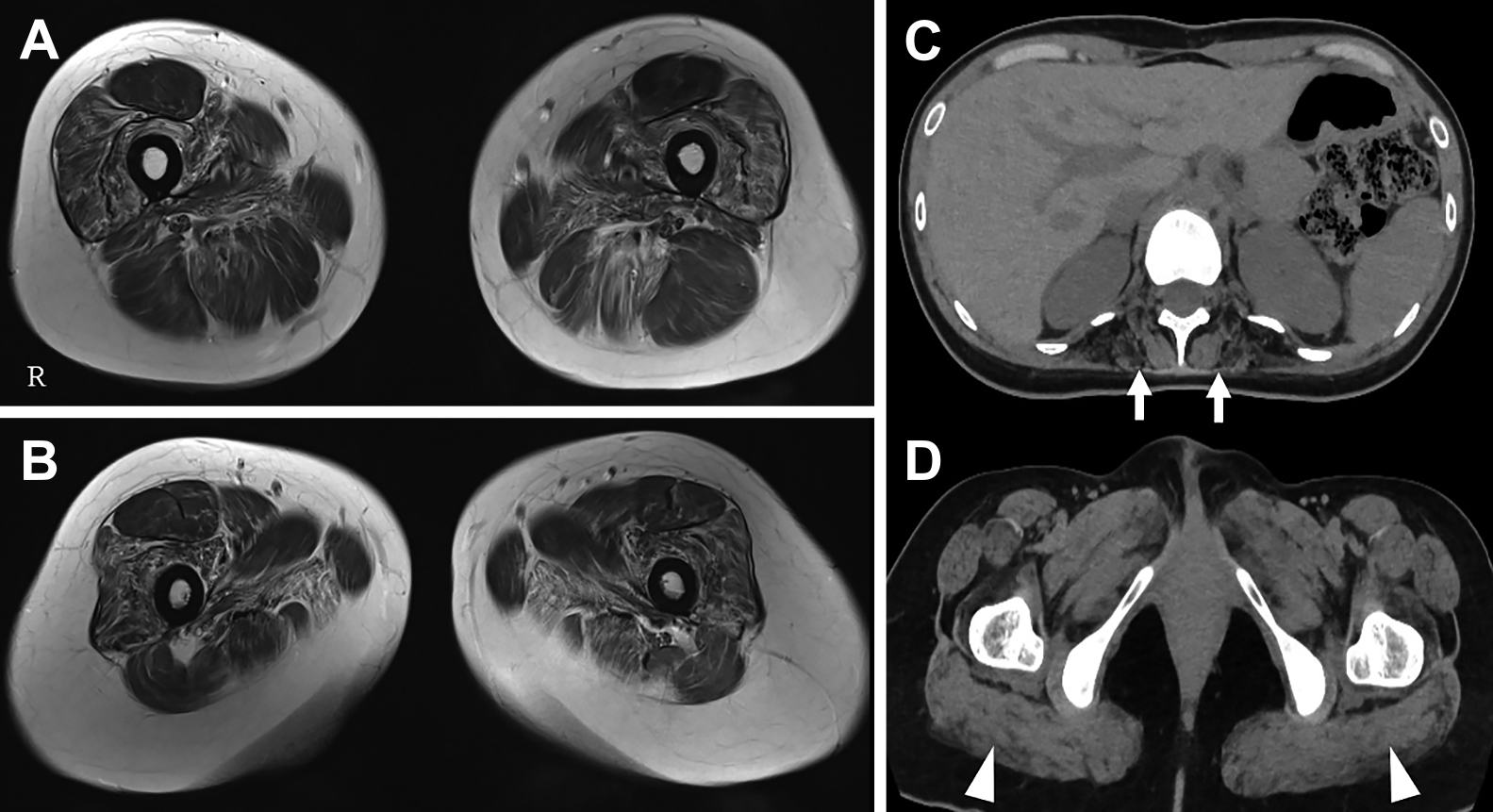
画像検査:大腿部MRIでは左優位に両側大腿~殿筋群にT2強調画像で脂肪変性を示唆する高信号と萎縮を認めた.特に外側広筋,中間広筋,半腱様筋,半膜様筋でめだった(Fig. 2A, B).全身CTでは傍脊柱筋や大殿筋,両下腿以遠の筋肉に萎縮・脂肪置換像を認めた(Fig. 2C, D).
入院後経過:本症例は,小児期発症の緩徐進行性の四肢・体幹の筋力低下を呈し,神経学的には下肢優位,遠位筋優位の筋力低下,深部腱反射消失,振動覚低下などの多発ニューロパチーを疑う所見を認めた.同時に体幹筋の筋力低下,CT・MRIでの体幹・四肢近位筋の筋萎縮,CK上昇を認めたことから,ミオパチーの存在も疑った.入院時の鑑別として,ミトコンドリア病,遠位型ミオパチー,遺伝性ニューロパチーなどを想定し,筋病理の確認と,遺伝子検査を提出した.
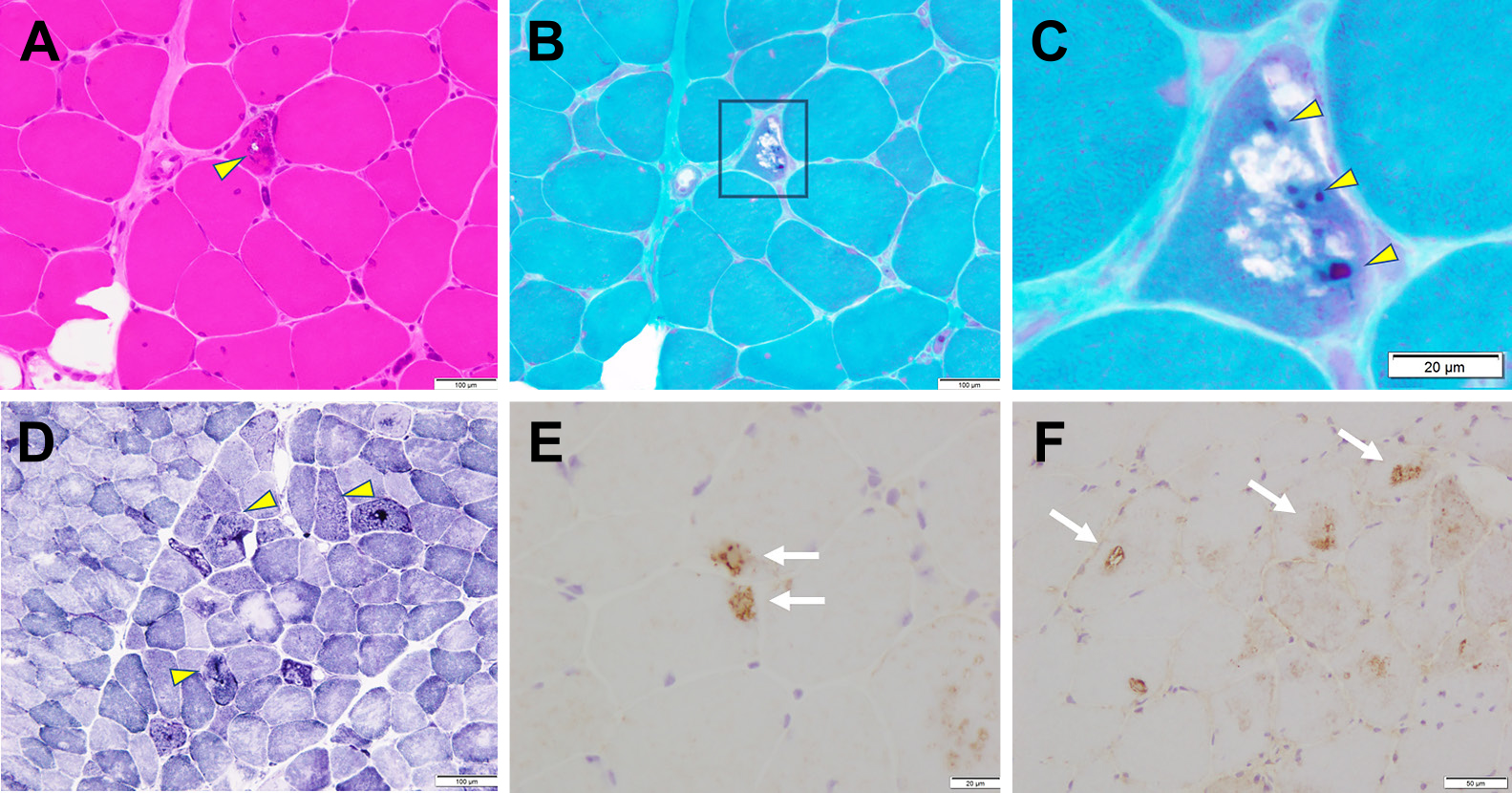
上腕二頭筋より筋生検を施行した.HE染色では筋線維の軽度の大小不同を認めるが,小角化線維や群集萎縮,pyknotic nuclear clampなどの神経原性変化を示唆する所見は認めなかった(Fig. 3A).筋線維タイプはモザイク状に分布し,偏りを認めなかった.HE染色,modified Gomori Trichrome染色でrimmed vacuole(RV)と,cytoplasmic bodyを認めた(Fig. 3A~C).RVは免疫組織化学染色でのP62,LC3沈着も認め,オートファジーの異常が示唆された(Fig. 3E, F).NADH-TR染色では筋原線維の軽度の乱れや虫食い像を認めた(Fig. 3D).以上より,MFMに矛盾しない所見であった.
遺伝性ニューロパチーに対して当科で遺伝子解析を実施した.遺伝子検査は倫理申請の承認を得た上で,患者と両親より書面によるインフォームド・コンセントを得て解析を行った.103遺伝子を対象としたCMT関連遺伝子ターゲットパネル解析3)にて,BAG3遺伝子に既報告のヘテロ接合性変異[c.626C>T (p.Pro209Leu)]を同定した.以上より,臨床的,病理学的所見,遺伝学的に,末梢神経障害を伴うMFM6と診断確定した.
なお,両親に同変異は認めず,de novo変異であった.
考察
本症例は,近位筋や体幹の筋力低下を呈し,針筋電図および筋病理で筋原性変化が示唆された.さらに,下垂足を呈し,神経伝導検査にて軸索障害を主体とした運動感覚性ニューロパチーを合併していた.MFM6としては典型的であったが,19歳時に歩行可能で心筋障害を伴わないという点で,既報告と比較して軽症であった.
MFMは,筋病理により定義される疾患で,BAG3,デスミン,ミオチリン,フィラミンCなどのZ帯に存在する蛋白質をコードする遺伝子変異が原因となって筋原線維構造を破綻させる4).BAG3遺伝子変異によるMFMは,MFM6に分類される.
MFMの病態メカニズムは,まずZ帯構造を保つ分子の欠陥によるZ帯の脆弱化が起こり,筋の収縮・弛緩による機械的ストレスによりZ帯が破壊され,やがて筋は収縮能を消失し,変性したZ帯蛋白が沈着,ストレス反応蛋白や蛋白処理機構にかかわる分子が誘導されるという筋崩壊の過程が推定されている5).
BAG3遺伝子がコードするBAG3蛋白は,Hsp70(70 kDa heat shock protein)と相互作用し,その働きを補完する.Hsp70は分子シャペロンとして機能し,蛋白質の凝集やミスフォールディングの防止,輸送,集合や分解などに関与する.特に,ストレス傷害により構造異常を起こした蛋白質に介添えして蛋白質を修復あるいは分解を促進して細胞内のホメオスタシスを維持するchaperone-assisted selected autophagy(CASA)にも関与している6).BAG3蛋白は骨格筋および心筋で強く発現しており,BAG3遺伝子異常ではこのCASA機構が機能不全に陥ることで,特に機械的ストレスを受けやすい骨格筋や心筋が障害される.同様の機序で,末梢神経細胞も障害されると考えられている2).
MFM6の遺伝形式は常染色体顕性遺伝(AD)だが,本症例のようにP209L変異では孤発例の報告が多い.
MFMの診断は,臨床所見,筋電図,神経伝導検査,そして最も重要な筋病理によってなされる.なお,これらによりMFMと診断されても,原因となる遺伝子変異が同定される割合は50%である4).Mayo ClinicでMFMと診断されたうちの5%がBAG3遺伝子変異によるものであるが,本邦での報告はP470S変異の2症例にとどまり7),頻度は不明である.
MFM6の臨床像は,四肢および体幹の筋力低下,軸索型の末梢神経障害,心筋症,呼吸不全,膝と足の関節拘縮,側弯症,脊椎強直を特徴とする.小児期の症例は特に重篤であり,急速な進行と青年期初期の死亡を伴う可能性がある8).
Table 27)~16)に,過去に報告されたBAG3関連疾患16例と本症例の表現型をまとめた.P209L変異の報告が最も多く,発症年齢は10歳前後と若く重症で,P470S変異では30代以降の発症で軽症であった.心合併症は17例中14例と多く,7例で拘束型心筋症,5例で肥大型心筋症(うち3例は閉塞性肥大型心筋症),5例で心房細動やQT延長などの不整脈を認め,3例で心臓移植が施行されていた.呼吸不全は14例で認め,3例で人工呼吸器の導入を要した.転帰として,4例が20歳以下で死亡した.このように同一変異であっても症例間で表現型の多様性が認められるが,その機序は明らかにされていない.BAG3以外の遺伝的背景の違い(oligogenicity)やBAG3発現に影響を与えるエピゲノム制御機構の違いが表現型に影響を与えている可能性があり,今後の研究課題である.また,環境要因として,末梢神経や骨格筋への機械的ストレスが起こりやすい生活習慣(例えば,運動負荷量が多い)や,心筋へのストレス(例えば,心拍数が多い)なども表現型を修飾する一因と思われる.一方,診断時の年齢や罹病期間の影響も考慮する必要があり,本症例では診断時には心筋障害を呈していないが,今後の長期の経過にて心筋障害を含めた他の徴候が出現する可能性もあるため,定期的な経過観察が必要であろう.
Table 2
Clinical feature of patients with BAG3-related myopathy.
| Reference |
Onset age (Y) |
Initial symptoms |
Cardiac disorder |
Respiratory failure |
Skeletal deformation |
NCS |
EMG |
Outcome |
BAG3 mutation |
Inheritance |
| Odgerel et al., 20109) |
5 |
NR |
RCM |
+ |
NR |
NR |
NR |
Sudden death at 9Y |
P209L |
AD |
| Odgerel et al., 20109) |
12 |
NR |
HOCM, HT |
+ |
NR |
Ax |
Neu + Myo |
HT at 14Y, MV at 29Y |
P209L |
AD |
| Odgerel et al., 20109) |
12 |
M, C |
RCM |
+ |
S, RS |
Ax |
Myo |
Death at 20Y |
P209L |
SP |
| Odgerel et al., 20109) |
5 |
M |
HOCM, HT |
+ |
NR |
Ax |
Neu + Myo |
HT at 13Y, Death at 15Y |
P209L |
SP |
| Lee et al., 201210) |
6 |
M |
RCM |
NR |
RS |
Ax |
Neu |
Ambulant at 12Y |
P209L |
SP |
| D’Avila et al., 201611) |
11 |
M |
HCM, ARR |
+ |
RS |
Ax |
Neu |
Not ambulant |
P209L |
SP |
| Selcen et al., 20098) |
toddler |
M |
RCM, HT |
+ |
NR |
NR |
NR |
HT at 13Y |
P209L |
SP |
| Selcen et al., 20098) |
13 |
M |
HCM, ARR |
+ |
S, RS |
Ax + Dem |
Myo |
nocturnal NPPV at 14Y |
P209L |
SP |
| Selcen et al., 20098) |
7 |
M |
RCM |
+ |
S, RS, WS |
NR |
NR |
Death at 13Y |
P209L |
SP |
| Kostera-Pruszczyk et al., 201512) |
12 |
M |
RCM, LQT |
− |
RS |
Ax + Dem |
NR |
Not ambulant |
P209L |
SP |
| Konersman et al., 201513) |
NR |
C |
RCM |
NR |
RS |
Ax |
Neu |
Not ambulant |
P209L |
SP |
| Scarpini et al., 202114) |
4 |
C |
HOCM |
+ |
RS |
Ax |
NR |
Ambulant with assistance |
P209L |
SP |
| Kim et al., 201815) |
11 |
M |
— |
+ |
RS, WS |
Ax |
Myo |
NR |
P209L |
SP |
| Andersen et al., 201816) |
10 |
M |
— |
+ |
S, RS |
NR |
NR |
MV at 19Y |
P209L |
SP |
| Hamaguchi et al., 20207) |
34 |
M |
LQT, Af |
+ |
NR |
Ax |
Myo |
NR |
P470S |
AD |
| Hamaguchi et al., 20207) |
40 |
M |
LQT, Af |
+ |
NR |
NR |
NR |
Ambulant with cane |
P470S |
AD |
| 本症例 |
10 |
M |
— |
+ |
WS |
Ax |
Myo |
Ambulant at 19Y |
P209L |
SP |
Y: years, NR: not reported, RCM: restricted cardiomyopathy, HCM: hypertrophic cardiomyopathy, HOCM: hypertrophic obstructive cardiomyopathy, LQT: long QT syndrome, Af: atrial fibrillation, ARR: arrythmia, HT: heart transplant, S: scoliosis, RS: rigid spine, WS: winged scapula, NCS: nerve conduction study, Ax: axonal neuropathy, Dem: demyelinating neuropathy, EMG: electromyography, Neu: neurogenic change, Myo: myogenic change, MV: mechanical ventilation, SP: sporadic, AD: autosomal dominant.
P209L変異では歩行不能となった症例が多く,確認できる範囲内では,介助なしで歩行可能な症例としては本症例の19歳が最高齢であった.脊椎側弯症や強直脊椎などの脊柱変形を10例で認めた.本症例のように翼状肩甲を来す症例も3例いた.9例に遠位および近位筋の筋力低下を認めたが,近位筋だけや遠位筋だけの筋力低下を認める症例もいる.近位筋だけの筋力低下を認めたミオパチー単独のような症例でも,神経伝導検査では軸索型ニューロパチーの合併を認めている.12例に神経伝導検査にて軸索型ニューロパチーが確認されている.針筋電図が施行された10例中7例に筋原性変化,5例に神経原性変化を認め,うち2例が両者混合であった.遺伝形式としては,P209L変異の15例中13例が孤発例で,残り2例はADであった.P470S変異の2例はADであった.
なお,BAG3変異の既報告例には見られず,本症例にだけ見られた徴候として,低身長と外眼筋麻痺があった.低身長については,BAG3変異以外のMFM症例では数例の報告があることから17)18),BAG3変異との関連を疑ったが,低身長である母親には同変異を認めなかった.したがって,本症例の低身長は母由来のBAG3変異以外の遺伝的要因による形質の可能性が最も考えられた.外眼筋麻痺については,左優位の両側外転制限を認めたが,末梢神経障害による外転神経障害であるか,ミオパチーによる外直筋障害であるかは不明であった.MFMにおいて外眼筋麻痺の既報告はないが,本疾患の新規の神経徴候である可能性もあり,眼窩部MRIを含めたさらなる精査が必要である.
このようにBAG3関連疾患におけるgenotype-phenotype correlationは未だ不明点が多く,今後も症例の蓄積,病理学的評価,疾患モデル生物を用いた機能解析などが必要である.
治療法については,現時点では対症療法が中心となる.心筋症に対しては心臓移植,呼吸不全に対しては人工呼吸器,拘縮に対してはリハビリテーション,下垂足に対しては装具処方などを行う.また,先述した発症機序の通り,機械的ストレスへの脆弱性があるため,激しい運動などの身体活動は避けるよう指導する.本症例でも,外来で心筋障害をフォローしながら,短下肢装具を使用して筋力維持および拘縮予防を目的としたリハビリテーションを継続している.治療研究も進んでおり,モデル動物を用いた遺伝子治療や,投薬研究が試みられている.BAG3遺伝子変異を過剰発現するマウスモデルにてアデノ随伴ウイルスベクターを用いた遺伝子治療法を行い,成長遅延や心不全の臨床症状など,特徴的な表現型が強く緩和された19).また,メトホルミンのドラッグリポジショニングも試みられており,BAG3変異ゼブラフィッシュモデルの運動機能を改善させ,ヒト筋芽細胞においてオートファジーを刺激し,蛋白質凝集体を除去し,筋線維の崩壊を防ぐことが示されている20).
各国からの症例報告も集積しており,稀であった本疾患も少しずつ臨床像が明らかになってきている.ニューロミオパチーや心筋症,呼吸不全や脊椎変形を伴うという特徴を踏まえ,孤発性であっても疑って遺伝子検査を行い診断し,治療や生活指導を行う必要がある.
Acknowledgments
謝辞:入院診療を担当した鹿児島大学病院 循環器内科 向井聡志先生,外来診療を担当している県立大島病院 脳神経内科 川畑裕太郎先生に深謝致します.
Notes
本報告の要旨は,第239回日本神経学会九州地方会で発表し,会長推薦演題に選ばれた.
※著者全員に本論文に関連し,開示すべきCOI状態にある企業,組織,団体はいずれも有りません.
文献
- 1) Scarpini G, Valentino ML, Giannotta M, et al. BAG3-related myofibrillar myopathy: a further observation with cardiomyopathy at onset in pediatric age. Acta Myologica 2021:177-183.
- 2) Shy M, Rebelo AP, Feely SM, et al. Mutations in BAG3 cause adult onset Charcot-Marie-Tooth disease. J Neurol Neurosurg Psychiatry 2018;89:313-315.
- 3) 樋口雄二郎,髙嶋博.遺伝性ニューロパチーの遺伝子診断.医のあゆみ 2022;283:1038-1045.
- 4) Fichna JP, Maruszak A, Żekanowski C, et al. Myofibrillar myopathy in the genomic context. J Appl Genet 2018;59:431-439.
- 5) 中野智.筋原線維性ミオパチー.臨床神経 2012;52:1151-1153.
- 6) 伊藤要子.ヒートショックプロテイン(HSP70)の魅力.日温気物医誌 2014;77:222-226.
- 7) Hamaguchi M, Kokubun N, Inoue M, et al. A family with adult-onset myofibrillar myopathy with BAG3 mutation (P470S) presenting with axonal polyneuropathy. Neuromuscular Disorders 2020;30:727-731.
- 8) Selcen D, Muntoni F, Burton BK, et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol 2009;65:83-89.
- 9) Odgerel Z, Sarkozy A, Lee HS, et al. Inheritance patterns and phenotypic features of myofibrillar myopathy associed with a BAG3 mutation. Neuromuscul Disord 2010;20:438-442.
- 10) Lee HC, Cherk SW, Chan SK, et al. BAG3-related myofibrillar myopathy in a Chinese family. Clin Genet 2012;81:394-398.
- 11) D’Avila F, Meregalli M, Lupoli S, et al. Exome sequencing identifies variants in two genes encoding the LIM-proteins NRAP and FHL1 in an Italian patient with BAG3 myofibrillar myopathy. J Muscle Res Cell Motil 2016;37:101-115.
- 12) Kostera-Pruszczyk A, Suszek M, Ploski R, et al. BAG3-related myopathy, polyneuropathy and cardiomyopathy with long QT syndrome. J Muscle Res Cell Motil 2015;36:423-432.
- 13) Konersman CG, Bordini BJ, Scharer G, et al. BAG3 myofibrillar myopathy presenting with cardiomyopathy. Neuromuscular Disorders 2015;25:418-422.
- 14) Scarpini G, Valentino ML, Giannotta M, et al. BAG3-related myofibrillar myopathy: a further observation with cardiomyopathy at onset in pediatric age. ACTA MYOLOGICA 2021;177-183.
- 15) Kim SJ, Nam SH, Kanwal S, et al. BAG3 mutation in a patient with atypical phenotypes of myofibrillar myopathy and Charcot-Marie-Tooth disease. Genes Genomics 2018;40:1269-1277.
- 16) Andersen AG, Fornander F, Schroder HD, et al. BAG3 myopathy is not always associated with cardiomyopathy. Neuromuscul Disord 2018;28:798-801.
- 17) Potulska-Chromik A, Jędrzejowska M, Gos M, et al. Pathogenic mutations and putative phenotype-affecting variants in polish myofibrillar myopathy patients. J Clin Med 2021;10:914.
- 18) Kim JY, Jeong EH, Park KD, et al. Myofibrillar myopathy: a case report. J Pathol Transl Med 2010;44:426-430.
- 19) Kimura K, Ooms A, Graf-Riesen K, et al. Overexpression of human BAG3P209L in mice causes restrictive cardiomyopathy. Nature Communications 2021;12:3575.
- 20) Ruparelia AA, McKaige EA, Williams C, et al. Metformin rescues muscle function in BAG3 myofibrillar myopathy models. Autophagy 2021;17:2494-2510.