要旨
症例は48歳男性.慢性進行性の両上肢の脱髄性末梢神経障害に加えて,左側胸部から下肢の感覚障害を主徴とする急性脊髄炎を発症し,中枢・末梢連合脱髄症(combined central and peripheral demyelination,以下CCPDと略記)と診断した.血清の抗ミエリンオリゴデンドロサイト糖蛋白(myelin oligodendrocyte glycoprotein,以下MOGと略記)抗体・抗galactocerebroside IgG抗体・抗GM1 IgG抗体が陽性であった.ステロイドパルス療法と血漿交換療法で脊髄炎は改善し,プレドニゾロン内服により末梢神経障害の所見は緩徐に改善し各種抗体はほぼ陰転化したが,8カ月後に神経根炎で再発した.抗MOG抗体関連疾患は再発を繰り返して新たな免疫反応を惹起し,CCPDを生ずる可能性がある.
Abstract
A 48-year-old male was admitted to our hospital because of chronic progressive demyelination of the peripheral nerves of the upper limbs, as well as acute myelitis presenting with sensory disturbance from the left chest to the left leg. We established a diagnosis of combined central and peripheral demyelination (CCPD). The patient was positive for serum anti-myelin oligodendrocyte glycoprotein (MOG), anti-galactocerebroside IgG, and anti-GM1 IgG antibodies. Intravenous methylprednisolone therapy and plasma exchange improved myelitis, and the subsequent administration of oral prednisolone yielded a gradual improvement of the peripheral nerve damage with a mostly negative result for the antibodies. However, the patient experienced a relapse of radiculitis eight months later. Relapses of anti-MOG antibody-associated disease can provoke new immune reactions, leading to CCPD.
はじめに
中枢・末梢連合脱髄症(combined central and peripheral demyelination,以下CCPDと略記)は中枢神経系と末梢神経系の両方に脱髄を来す稀な疾患である.2015年にOgataらによりその特徴がまとめられ,診断基準案が提案された1).抗neurofascin抗体などの病態への関与が指摘される一方で2),近年抗ミエリンオリゴデンドロサイト糖蛋白(myelin oligodendrocyte glycoprotein,以下MOGと略記)抗体陽性のCCPDが複数例報告されている3)~7).抗MOG抗体に加え抗galactocerebroside(Gal-C)抗体と抗GM1抗体が陽性のCCPDの症例を経験したため,貴重な症例と考え報告する.
症例
症例:48歳 男性
主訴:左側胸部から下肢にかけての違和感
既往歴:胃潰瘍(10歳代).
家族歴:類症なし.
生活歴:喫煙 5本/日,26年間.飲酒 なし.
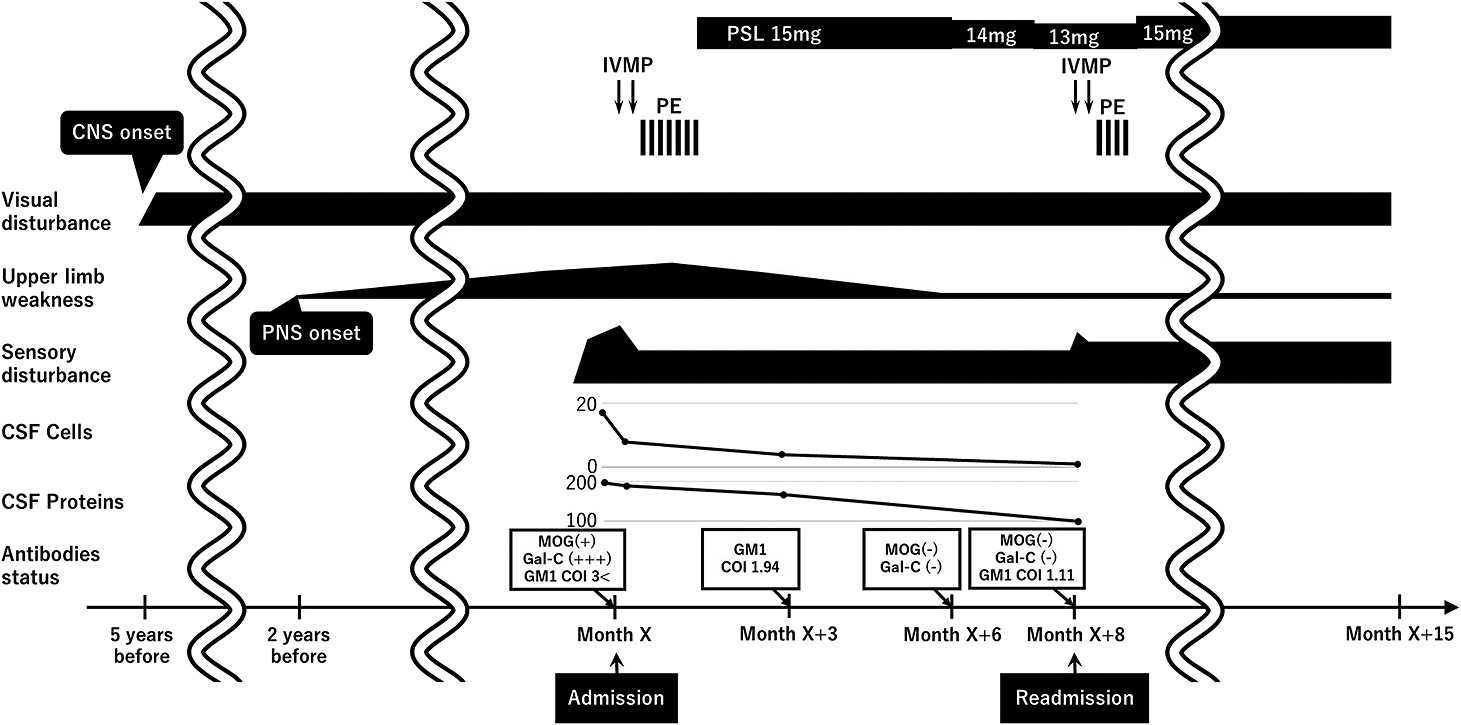
現病歴:5年前に急性に両眼の視力低下が出現し,同程度のままで残存した.自動車運転は可能で生活に不自由はなかった.1年前頃からゴルフのドライバーショットで球に当たらなくなり,両上肢の筋力低下が進行していると感じた.X月上旬に左側胸部の違和感(ぽかぽかと温かい感じ)が始まり,数日間で左下肢まで違和感が拡大したため来院し,精査目的に入院した.入院前数カ月以内に発熱や呼吸器症状はなかった.
入院時身体所見:身長165.0 cm,体重52.0 kg.血圧119/84 mmHg,脈拍68回/分・整.呼吸数12回/分,SpO2 97%(室内気下).体温36.2°C.胸部の聴診や腹部の触診に異常はなく,脊柱叩打痛はなかった.意識レベルはGCS E4V5M6であった.両眼の視力障害以外に脳神経に異常はなかった.徒手筋力検査は四肢でMMT5であり,握力は右が30 kg,左が35 kg(右利きで病前は両側40 kg以上)であった.鼻指鼻試験と踵膝試験で失調を認めなかった.感覚系は左Th4高位以下で末梢優位の触覚低下と痛覚脱失を認めた.振動覚は左右差なく正常であった.背部に放散するLhermitte徴候が陽性であった.深部腱反射において,上腕二頭筋腱反射,腕橈骨筋腱反射,上腕三頭筋腱反射とアキレス腱反射は正常で,膝蓋腱反射は軽度減弱していたが左右差はなかった.Babinski反射は右で背屈,左は底屈していた.
検査所見:血算や一般生化学検査に異常は認めなかった.可溶性インターロイキン2レセプターは176 U/ml(基準値157~474),アンジオテンシンI転換酵素は13.7 U/l(8.3~21.4),HbA1cは5.6%でビタミンB1,B12,葉酸は正常値であった.自己抗体検査では抗核抗体,抗SS-A・抗SS-B抗体,抗MPO-ANCA・抗PR3-ANCA抗体,抗アクアポリン4抗体(ELISA法),抗NF155抗体,抗CNTN1抗体はいずれも陰性で,抗MOG抗体(cell-based assay(CBA)法)が陽性であった.抗ガングリオシド抗体(GM1,GM2,GD1a,GD1b,GT1a,GQ1b,GA-1)のうち,抗GM1 IgG抗体がCOI 3以上(0.7未満)と陽性で,抗Gal-C IgG抗体も(+++)と陽性であった.Mycoplasma pneumoniae抗体は陰性であった.血清蛋白分画ではM蛋白を認めなかった.髄液所見は無色透明で初圧145 mmH2O,細胞数17/μl(単核球77%,多形核球23%),蛋白197 mg/dl,糖51 mg/dl(同時血糖78 mg/dl)であり,IgG index 0.88であった.オリゴクローナルバンド(oligoclonal bands,以下OCBと略記)は陰性で,ミエリン塩基性蛋白127 pg/ml(102未満)であった.髄液中の抗MOG抗体がCBA法で陽性であった.眼底検査では異常がなかった.頭部MRI画像では左側脳室近傍に造影効果を伴わない最大径約10 mmのT2高信号域を認め,明らかな視神経の信号変化を認めなかった.頸椎MRIにてC3~7高位にかけて中心灰白質から右方に広がるT2高信号域を認め,C4~5高位では造影効果を呈した(Fig. 1).神経伝導検査(nerve conduction study,以下NCSと略記)では両側の正中,尺骨神経において遠位潜時の延長,運動神経伝導速度の低下,複合筋活動電位(compound motor action potential,以下CMAPと略記)振幅の軽度低下を認めた.F波は両側正中,尺骨,後脛骨神経において潜時の延長,右正中,右尺骨,左後脛骨神経において出現率の軽度低下を認めた.下肢では両側後脛骨神経にてCMAP振幅の軽度低下を認めた(Table 1).両側正中神経で慢性炎症性脱髄性多発根神経炎の基準を満たした8).視覚誘発電位(visual evoked potential,以下VEPと略記)では片眼刺激・半視野刺激にて両側で潜時の延長を認めた.聴性脳幹反応は正常であった.

Table 1
Nerve conduction study (NCS).
|
Nerve |
On admission
(Month X) |
Month X+1 |
Month X+4 |
On readmission†
Month X+8 |
| MCV (m/s) |
Lt Median |
46.9 |
51.4 |
50.5 |
49.8 |
|
Lt Ulnar |
43.6 |
46.1 |
50.0 |
49.0 |
|
Lt P. Tib |
43.6 |
47.2 |
44.7 |
40.2 |
| DL (ms) |
Lt Median |
7.5 |
6.9 |
4.8 |
4.3 |
|
Lt Ulnar |
4.3 |
4.4 |
3.4 |
3.2 |
|
Lt P. Tib |
4.7 |
7.1 |
3.8 |
5.0 |
| CMAP amplitude (mV) |
Lt Median |
4.2 |
3.5 |
7.3 |
10.5 |
|
Lt Ulnar |
6.5 |
8.0 |
9.7 |
11.8 |
|
Lt P. Tib |
6.4 |
9.6 |
8.8 |
11.9 |
| SCV (m/s) |
Lt Median |
42.8 |
42.2 |
48.2 |
46.1 |
|
Lt Ulnar |
40.5 |
46.5 |
46.5 |
47.0 |
|
Lt Sural |
49.6 |
42.2 |
51.2 |
41.4 |
| SNAP amplitude (μV) |
Lt Median |
3.2 |
5.7 |
20.5 |
18.4 |
|
Lt Ulnar |
2.3 |
7.4 |
14.0 |
15.0 |
|
Lt Sural |
22.9 |
23.1 |
21.4 |
21.5 |
| F-occ. (%) |
Lt Median |
87 |
50 |
62 |
62 |
|
Lt P. Tib |
93 |
100 |
100 |
100 |
| Max FWCV (m/s) |
Lt Median |
46.6 |
53.3 |
56.8 |
56.5 |
|
Lt P. Tib |
37.6 |
38.0 |
42.4 |
36.9 |
| Min F-latency (ms) |
Lt Median |
37.7 |
34.9 |
30.5 |
30.2 |
|
Lt P. Tib |
51.5 |
55.4 |
49.0 |
53.7 |
† On readmission, NCS was performed before treatment. MCV, motor conduction velocity; DL, distal latency; CMAP, compound motor action potential; SCV, sensory conduction velocity; SNAP, sensory nerve action potential; F-occ., F-wave occurrence; max FWCV, maximum F-wave conduction velocity; min F-latency, minimum F-wave latency; Lt, left; P. Tib, post tibial.
入院後経過:急性期治療としてステロイドパルス療法(intravenous methylprednisolone pulse therapy(IVMP)1 g/日,3日間)を2クール施行し,左側胸部以下の感覚障害は軽度の改善を認め,頸髄MRIの造影効果は消失したが,両上肢の筋力低下は改善しなかった.次に単純血漿交換療法(plasma exchange,以下PEと略記)を7回施行し,握力が左 38 kg,右 37 kgに改善して感覚障害も軽度の改善を認めた.プレドニゾロン(prednisolone,以下PSLと略記)15 mg/日の内服を開始して入院第42病日に自宅へ退院した.退院後は両上肢の筋力低下とNCS,髄液の所見は改善傾向となった(Table 1, 2).X+3~6月に血清の抗MOG抗体と抗糖脂質抗体を再検査すると抗GM1 IgG抗体のみ弱陽性(COI 1.94)であったが他は全て陰性であった.内服のPSLを13 mg/日まで漸減したところ,X+8月に左大腿背外側のじんじんとした痺れ感が出現し,1週間の経過で増悪したため入院した.入院時に左側でLasègue徴候が陽性であった.抗GM1 IgG抗体はCOI 1.11であった.脊髄MRIで馬尾の肥厚と造影効果を認め,神経根炎と診断した(Fig. 1).IVMPを2クール施行し症状の悪化は止まったが馬尾の造影効果は残存した.PEを4回と免疫グロブリン大量静注療法(IVIg)を施行し,馬尾の造影効果は消失したが,感覚障害は残存した.ADLは自立を維持し,PSL 15 mg/日の内服を継続している(Fig. 2).
Table 2
Reported patients of anti-MOG antibody positive combined central and peripheral demyelination (CCPD).
Age and sex
on admission |
CNS & PNS onset |
CNS lesions |
PNS lesions |
Treatment |
Clinical course |
| Acute |
Maintenance |
Central |
Peripheral |
| 18 M3) |
Simultaneous |
Supra/Infra-tentorial,
C5–T1, T10–11, conus |
Lt limbs,
cauda equina |
IV steroid |
N/A |
CR |
CR |
| 51 M4) |
Different (C→P) |
T12-conus |
Rt tibial,
cauda equina |
IVMP
PSL 60 mg/day |
AZA
PSL 10 mg/day |
Recurrences |
N/A |
| 46 M5) |
Different (C→P→C+P) |
ON |
Four limbs,
cauda equina |
IVMP, PE |
PSL 15 mg/day |
CR |
PR |
| 32 F6) |
Simultaneous |
PVWM, pons,
C4, C7, T9 |
Four limbs,
cauda equina |
IVIg, IVMP |
N/A |
Recurrence |
Recurrences |
| 9 F7) |
Simultaneous |
T3–T10, L1/conus |
Four limbs,
nerve roots |
IVMP, IVIg |
DEX |
PR |
| 34 M7) |
Simultaneous |
Leptomeningeal, C2 |
Lower limbs,
cauda equina |
DEX, IVIg |
N/A |
CR |
| 26 F7) |
Different (P→C) |
R ON |
Rt median |
Monthly IVIg for 2 years |
CR |
| Present case |
Different (C→P) |
ON, PVWM, C3–7 |
Upper limbs,
cauda equina |
IVMP, PE |
PSL 15 mg/day |
PR |
Recurrence |
PSL, prednisolone; AZA, azathioprine; IVIg, intravenous immunoglobulins; IVMP, high-dose glucocorticoid; PVWM, periventricular white matter. ON, optic neuritis; DRG, dorsal root ganglion; BBD, bladder and bowel dysfunction; CR, complete resolution; PR, partial resolution.
考察
本症例は急性発症の感覚障害で受診し,MRIで3椎体以上の脊髄病変を認めて脊髄炎と診断した.しかしながら慢性進行性の両上肢筋力低下の病歴,深部腱反射の亢進がないこと,NCSにおける両上肢の遠位優位の脱髄所見を認めたことからCCPDを疑った.CCPDは中枢・末梢神経系の両者に脱髄を来す稀な疾患である.Ogataらによる診断基準案としては① MRIやVEPでの中枢神経病変,② NCSで二つ以上の末梢神経における脱髄所見,③ 感染症や既存の炎症性疾患など明らかな原因の除外,の3項目を満たすこととされる1).本症例はこの3項目を満たし,CCPDと診断した.本症例では急性期の脊髄病変に加え,MRIで造影効果のない脳病変が存在し,病歴とVEPから視神経炎の既往もあり,抗MOG抗体関連の再発寛解型の中枢神経障害を繰り返していたと考えた.一方で両上肢の筋力低下は慢性経過の末梢神経障害によるものと考えた.Ogataらの同報告によると,中枢神経系と末梢神経系の症状について,同時期と異時期の発症がそれぞれ21,79%であり,異時期の発症例のうち再発寛解型が66%で慢性進行型が24%であった.深部腱反射の低下は65%で認め,髄液検査では蛋白上昇が83%にみられたが細胞数増多やOCB陽性,IgG indexの上昇は10~20%程度と少なかった.MRIでは脊髄病変を75%に認めるが,本症例のような脊髄長大病変は7.5~21%とされる1)9).なお,本症例において脱髄性末梢神経障害は上肢近位部と遠位部にそれぞれ認めた.抗ミエリン随伴性糖蛋白抗体は測定していないが,M蛋白は認めなかった.精査にて抗MOG抗体,抗Gal-C IgG抗体,抗GM1 IgG抗体の陽性が判明した.これら複数の抗体が陽性となったCCPDの例は報告がない.Ogataらの報告では80%以上に奏功したIVMPとPEを本症例でも施行した1).
本症例では脊髄炎発症時の血清,髄液において抗MOG抗体が陽性であった.MOGはヒトでは中枢神経系の髄鞘にのみ存在し,抗MOG抗体関連疾患(MOG antibody-associated disease,以下MOGADと略記)は中枢神経系の脱髄疾患を呈する10)11).しかしながら,MOGADの免疫反応は末梢の免疫系にて誘導されることから12),末梢神経の脱髄に関連し得ると考えた.2018年に抗MOG抗体陽性のCCPD症例が初めて報告され3),CCPDの診断基準案を満たす症例は7例報告されている(Table 2)3)~7).この7例のうち3例が異時期発症であった.7例のうち6例で馬尾の造影効果を認め,抗MOG抗体陽性のCCPDに特徴的な所見と考えた.抗MOG抗体陽性のCCPDの病態仮説として,MOGが髄腔から末梢神経に移行して自己免疫を賦活するという説や他の霊長類と同様にヒトでもMOGが末梢神経にも存在するという説,単一の自己免疫病態が中枢・末梢神経系の両方に炎症を起こしているという説などが提案されている3).また,本症例では血清の抗Gal-C IgG抗体と抗GM1 IgG抗体も陽性であった.自己免疫疾患において複数の自己抗原に対する免疫応答に関与する現象としてエピトープスプレディングが示唆されている13)14).MOGによって誘導された実験的自己免疫性脳脊髄炎モデルマウスでこの現象が報告されており,本症例でも関与していると考えた15).
Gal-Cは中枢・末梢神経系の両方の髄鞘に存在するスフィンゴ糖脂質で,抗Gal-C抗体は多発性硬化症や急性散在性脳脊髄炎(acute disseminated encephalomyelitis,以下ADEMと略記),Guillain–Barré症候群(Guillain–Barré syndrome,以下GBSと略記)などの脱髄疾患の一部で検出され,特にIgG抗体はGBSやMiller Fisher症候群の一部にみられる16).本邦から抗Gal-C IgG抗体単独陽性のCCPDでIVMPが著効した症例報告がある17).本症例ではIVMPのみの効果は限定的であったが,発症から治療までの期間が長かったことが影響したかもしれない.なお,抗Gal-C抗体はMycoplasma pneumoniae感染との関連が示唆されているが,本症例では先行感染はなく,入院時のMycoplasma pneumoniae抗体は陰性であった.また,抗Gal-C抗体は大脳,脊髄,神経根,末梢神経に脱髄を来すEncephalomyeloradiculoneuropathy(EMRN)に関与するとの報告があり18),本症例もEMRNとの異同が問題になるが,EMRNとCCPDとは鑑別の基準がなく,現時点では不明である19).
抗GM1 IgG抗体は軸索型のGBSの病因となる抗体であり,ADEMでの陽性報告例はあるがその病態は不明である20).本症例は両者とも異なっており,抗GM1 IgG抗体の病的意義は不明である.なお,CCPDで多く報告がある抗NF155抗体は本症例では陰性であった1)21).
本症例はMOGADの一部であり,中枢と末梢で別々の病態が偶然併発したと考えるより,まず末梢で免疫誘導されて中枢神経障害を発症し,その再発を繰り返すことで新たな免疫誘導を引き起こし,ついに末梢神経障害を惹起したと考えた.また,抗MOG抗体陽性CCPDの7例中4例で神経障害の残存や再発がみられ(Table 2),うち1例では抗MOG抗体が持続陽性であった6).MOGADのうちCCPDでは疾患活動性が高いことが示唆され,注意が必要である3)~7).末梢神経障害の病態としては近位部及び遠位部の脱髄を呈しており,血液神経関門を欠く部位を標的とした液性免疫の関与が考えられ22),PEやIVIgの有効性と合致する.MOGADが頻回に再発することでCCPDに発展するのであれば,MOGADは初発時から可能な限り再発を予防することが一層重要である.さらに,CCPDを発症した場合は疾患活動性が高いと考えられ,MOGADの液性免疫を標的とした治療法も検討すべきかもしれない23)~25).
CCPDは経過や所見が多様で初発症状をとらえられない症例が存在する.CCPDと診断した場合,既知の中枢/末梢神経脱髄疾患の抗体を検索することで治療選択の一助となる可能性があるが,抗MOG抗体陽性例はその活動性に注意が必要である.MOGADの再発を繰り返すことで新たな免疫誘導が惹起される可能性があるため,強力な再発予防と慎重な観察が必要である.MOGADで末梢神経障害を合併しやすい症例の特徴は不明であり,その病態解明に向けて今後のさらなる症例の蓄積と検討が待たれる.
Acknowledgments
謝辞:血清と髄液の抗MOG抗体を測定頂きました東北大学大学院医学系研究科神経内科学講座 高橋利幸先生,血清の抗ガングリオシド抗体を測定頂きました近畿大学の楠進先生,血清の抗NF155抗体と抗CNTN1抗体を測定頂きました名古屋大学の深見祐樹先生に深謝致します.
Notes
本報告の要旨は,第162回日本神経学会東海・北陸地方会で発表し,会長推薦演題に選ばれた.
※著者全員に本論文に関連し,開示すべきCOI状態にある企業,組織,団体はいずれも有りません.
文献
- 1) Ogata H, Matsuse D, Yamasaki R, et al. A nationwide survey of combined central and peripheral demyelination in Japan. J Neurol Neurosurg Psychiatry 2016;87:29-36.
- 2) Kawamura N, Yamasaki R, Yonekawa T, et al. Anti-neurofascin antibody in patients with combined central and peripheral demyelination. Neurology 2013;81:714-722.
- 3) Vazquez Do Campo R, Stephens A, Marin Collazo IV, et al. MOG antibodies in combined central and peripheral demyelination syndromes. Neurol Neuroimmunol Neuroinflamm 2018;5:e503.
- 4) Sundaram S, Nair SS, Jaganmohan D, et al. Relapsing lumbosacral myeloradiculitis: an unusual presentation of MOG antibody disease. Mult Scler 2020;26:509-511.
- 5) Shima T, Tsujino A. MOG antibody-related disease with recurrent optic neuritis and sensory polyradiculoneuropathy: a case report. Mult Scler Relat Disord 2020;46:102597.
- 6) Nakamura T, Kaneko K, Watanabe G, et al. Myelin oligodendrocyte glycoprotein-IgG-positive, steroid-responsive combined central and peripheral demyelination with recurrent peripheral neuropathy. Neurol Sci 2021;42:1135-1138.
- 7) Rinaldi S, Davies A, Fehmi J, et al. Overlapping central and peripheral nervous system syndromes in MOG antibody–associated disorders. Neurol Neuroimmunol Neuroinflamm 2021;8:e924.
- 8) Van den Bergh PYK, Doorn PA, Hadden RDM, et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint Task Force-Second revision. J Peripheral Nervous Sys 2021;26:242-268.
- 9) Cortese A, Franciotta D, Alfonsi E, et al. Combined central and peripheral demyelination: clinical features, diagnostic findings, and treatment. J Neurol Sci 2016;363:182-187.
- 10) Takai Y, Misu T, Kaneko K, et al. Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study. Brain 2020;143:1431-1446.
- 11) Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol 2019;15:89-102.
- 12) Marignier R, Hacohen Y, Cobo-Calvo A, et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol 2021;20:762-772.
- 13) Lehmann PV, Forsthuber T, Miller A, et al. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature 1992;358:155-157.
- 14) Cornaby C, Gibbons L, Mayhew V, et al. B cell epitope spreading: mechanisms and contribution to autoimmune diseases. Immunol Lett 2015;163:56-68.
- 15) Flytzani S, Guerreiro-Cacais AO, N’diaye M, et al. MOG-induced experimental autoimmune encephalomyelitis in the rat species triggers anti-neurofascin antibody response that is genetically regulated. J Neuroinflammation 2015;12:194.
- 16) Kuwahara M, Samukawa M, Ikeda T, et al. Characterization of the neurological diseases associated with Mycoplasma pneumoniae infection and anti-glycolipid antibodies. J Neurol 2017;264:467-475.
- 17) Hoshino M, Suzuki Y, Akiyama H, et al. Efficacy of high-dose steroid pulse therapy for anti-galactocerebroside antibody-positive combined central and peripheral demyelination. Rinsho Shinkeigaku 2017;57:747-752.
- 18) Shima S, Kawamura N, Ishikawa T, et al. Anti-neutral glycolipid antibodies in encephalomyeloradiculoneuropathy. Neurology 2014;82:114-118.
- 19) Saito K, Toru S, Shima S, et al. Anti-neutral glycolipids antibody-positive combined central and peripheral demyelination mimicking encephalomyeloradiculoneuropathy phenotype. Clin Neurol Neurosurg 2018;172:90-92.
- 20) Young JW, Mason DF, Taylor BV. Acute inflammatory encephalomyelitis following Campylobacter enteritis associated with high titre antiganglioside GM1 IgG antibodies. J Clin Neurosci 2009;16:597-598.
- 21) Devaux JJ, Miura Y, Fukami Y, et al. Neurofascin-155 IgG4 in chronic inflammatory demyelinating polyneuropathy. Neurology 2016;86:800-807.
- 22) Koike H, Katsuno M. Pathophysiology of chronic inflammatory demyelinating polyneuropathy: insights into classification and therapeutic strategy. Neurol Ther 2020;9:213-227.
- 23) Chen JJ, Flanagan EP, Bhatti MT, et al. Steroid-sparing maintenance immunotherapy for MOG-IgG associated disorder. Neurology 2020;95:e111-e120.
- 24) Kuwabara S, Isose S, Mori M, et al. Different electrophysiological profiles and treatment response in ‘typical’ and ‘atypical’ chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry 2015;86:1054-1059.
- 25) Chen JJ, Huda S, Hacohen Y, et al. Association of maintenance intravenous immunoglobulin with prevention of relapse in adult myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol 2022;79:518-525.
