要旨
症例は36歳男性.35歳時より感覚障害を伴わない左母指筋力低下,左母指球筋と左第一背側骨間筋の萎縮が出現した.神経伝導検査で左正中神経に複合筋活動電位振幅低下とF波出現率低下,針筋電図検査で左短母指外転筋に陽性鋭波を認めた.頭部MRIで両側小脳半球は萎縮し,母方祖母と母の兄弟が脊髄小脳変性症と判明した.その後,両下肢失調が現れ遺伝子検査でATXN2遺伝子CAGリピート数の伸長(19/39)を認めた.下位運動ニューロン障害が初発症状の脊髄小脳失調症2型と診断した.ATXN2遺伝子リピート伸長を示す同一家系内で小脳失調症と運動ニューロン障害を呈することが報告されており興味ある症例と考え報告する.
Abstract
A 36-year-old man has developed weakness of left thumb and atrophy of left thenar muscle and left first dorsal interosseous muscle without sensory disturbance for a year. A nerve conduction study revealed decreases in the amplitude of compound muscle action potentials and occurrence of F-waves on left medial nerve. Needle electromyography examination revealed positive sharp waves and later recruited motor units on left abductor pollicis brevis muscle. Brain MRI showed atrophy of bilateral cerebellar hemisphere. His grandmother and his two uncles have been diagnosed as spinocerebellar degeneration. After discharge, he developed bilateral lower limb ataxia. Genetic analysis showed heterozygous CAG repeat expansion (19/39) in ATXN2 gene, being diagnosed as spinocerebellar ataxia 2 (SCA2). A previous report has shown that motor neuron involvement is recognized as part of SCA2 in the same pedigree with full CAG repeat expansions in ATXN2 gene. We here report the patient with lower motor neuron involvement as an initial symptom of SCA2.
はじめに
脊髄小脳失調症2型(spinocerebellar ataxia 2,以下SCA2と略記)は,第12染色体長腕(12q23-24)のATXN2遺伝子におけるCAGリピートの異常伸長により発症する遺伝性脊髄小脳変性症である1)2).通常は小脳性運動失調で発症するが,小脳性運動失調以外に多彩な神経症状を認めることが知られており,6~7割の症例で末梢神経障害を呈する3)4).約1割に下位運動ニューロン障害がみられるが,初発症状となる例は少ない3).今回,私たちは下位運動ニューロン障害を初発症状とするSCA2の1例を経験したので報告する.
症例
症例:36歳,男性
主訴:左母指の脱力
既往歴:特記すべき事項なし.
家族歴(Fig. 1):母親(II-2)は神経症状の有無に関して詳細不明であったが,小脳萎縮を指摘されたことがあると患者本人より聴取した.母方の叔父2人(II-3,II-4)はいずれも30歳代で脊髄小脳変性症(spinocerebellar degeneration,以下SCDと略記)と診断された.母方の従兄弟(III-3)は20歳代でSCDと診断された.母方の祖母(I-2)は55歳で死亡した.詳細は不明であるがSCDを指摘されたことがある.全員沖縄県の出身である.
生活歴:飲酒なし,喫煙なし.出身は沖縄県で,職場でトルエンの曝露歴があった.
内服歴:市販の複合ビタミン剤を服用していた.
現病歴:35歳時に左母指の筋力低下を自覚し,当科外来を受診した.左母指球筋に萎縮を認めたが,左手に感覚障害を認めなかった.運動神経伝導検査では,左正中神経の遠位潜時延長と,両側正中神経のF波導出頻度低下(右13%,左19%)を認めた.神経伝導速度は正常範囲であった.感覚神経伝導検査では,左正中神経に特記すべき所見を認めなかった.左手関節MRIで有鈎骨レベルでの正中神経扁平化を認め,左正中運動神経の遠位潜時延長の原因と考えた.True neurogenic thoracic outlet syndromeを示唆するような左内側前腕皮神経の感覚神経伝導の異常は見られなかった.原因不明のまま経過観察としていたが,その後も緩徐に筋力低下が進行したため,36歳時に当科入院した.
一般身体所見:身長170.8 cm,体重73.3 kg.体温36.7°C.脈拍72回/分(整).血圧134/74 mmHg.SpO2 98%(room air).
神経学的所見:意識清明であった.眼振,眼球運動異常,構音障害は認めず,脳神経系に異常所見を認めなかった.徒手筋力検査では左短母指外転筋が3,長母指屈筋・第一背側骨間筋・小指外転筋が左で4であった.左母指球筋及び左第一背側骨間筋に萎縮を認める一方で,左小指球筋に萎縮はなくSplit handを呈していた5).筋線維束性収縮は観察されず,四肢腱反射は正常で病的反射はなかった.感覚障害は認めなかった.指鼻試験,膝踵試験,反復拮抗運動で明らかな異常はなかった.歩行は正常であった.入院時点で小脳失調所見は明らかでなかった.
入院時検査所見:全血球計算は正常で,血液生化学検査ではCK 450 U/lと上昇を認めた.抗核抗体,抗SS-A抗体,MPO-ANCA,PR3-ANCA,抗ARS抗体,抗アセチルコリンレセプター抗体は陰性で,脳脊髄液検査は異常を認めなかった.心電図,胸部X線で明らかな異常は認めなかった.運動神経伝導検査では,左正中神経の複合筋活動電位は導出されず,左尺骨・腓骨神経において振幅低下を認め,右正中神経のF波導出頻度低下と左尺骨神経のF波消失も認めた.左尺骨・腓骨神経,右正中神経の伝導速度は正常範囲であった(Fig. 2).感覚神経伝導検査は正常範囲であった.針筋電図検査では左上腕二頭筋,左総指伸筋に動員遅延,持続時間延長といった神経原性変化を認め,左短母指外転筋に陽性鋭波と動員遅延を認めた.右短母指外転筋と右僧帽筋の反復刺激検査で異常は認めなかった.呼吸機能検査・内視鏡下嚥下機能検査では特記すべき異常を認めなかった.FISH法にてPMP22遺伝子に欠失や重複は認められなかった.
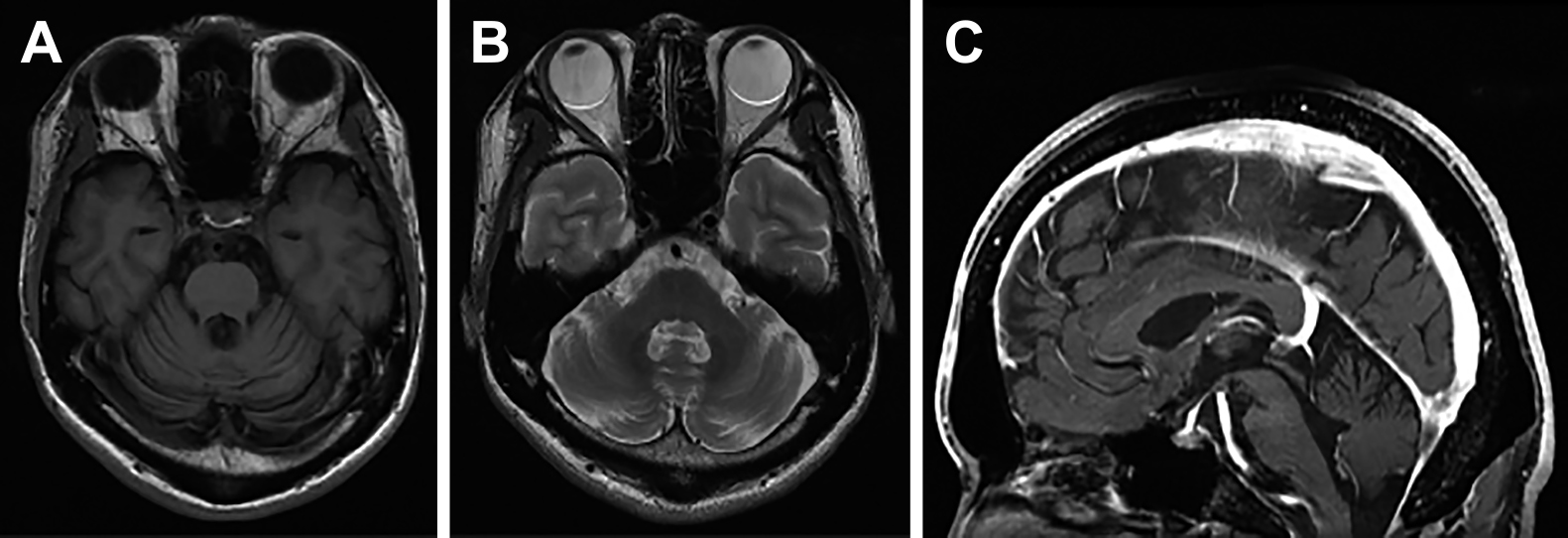
画像検査:頭部MRIで両側小脳半球の萎縮を認めた.中脳被蓋及び中小脳脚は保たれ,中小脳脚の信号変化やHot cross bun signは認めなかったが,橋尾側底部に軽度の萎縮を認めた(Fig. 3).脊椎単純造影MRIでは髄内に異常信号を認めなかった.
経過:左正中神経の支配筋にみられた筋力低下と筋萎縮は,その支配領域に感覚障害を認めない,神経伝導検査でF波導出不良が先行した後に複合筋活動電位低下が出現した,運動神経伝導速度は正常範囲で感覚神経伝導の異常を認めない,針筋電図検査で脱神経変化を伴う神経原性変化を認めるといった所見を呈し,下位運動ニューロン障害と考えられた.この時点で小脳性運動失調は明らかではなかったが,小脳萎縮とSCDの家族歴より,遺伝性脊髄小脳変性症に伴う下位運動ニューロン障害の可能性が考えられた.退院2か月後,膝踵試験で測定過大傾向と継ぎ足歩行での不安定性の出現を認めた.ご本人の同意を得て遺伝子検査を施行した.PCRフラグメント解析で歯状核赤核淡蒼球ルイ体萎縮症,ハンチントン病,SCA1/2/3/6/7/8/12/17/31の原因遺伝子,およびRepeat-primed PCR法でSCA36の原因遺伝子におけるリピート伸長変異について解析したところ,SCA2の原因遺伝子であるATXN2遺伝子においてCAGリピート伸長を認めた.それ以外の遺伝子に病的と考えられるリピート伸長変異はなかった.GeneScan(Applied Biosystems, japan)を活用したATXN2遺伝子のDNAフラグメント解析でCAGリピート数を19/39と算出し,ヘテロ接合性のリピート伸長変異を確定した.以上よりSCA2と診断した.タルチレリン水和物の内服を開始した.その後,左尺骨運動神経,右正中運動神経の複合筋活動電位が導出不可となり,右尺骨運動神経と両側橈骨運動神経の振幅低下があり,下位運動ニューロン障害の進行を認めた.
考察
SCA2はataxin 2をコードする染色体12q24上のATXN2遺伝子におけるCAG反復配列の異常伸長により生じるポリグルタミン病である.30歳代の発症が多く,発症後10~15年の経過で死亡する6).初発症状としては小脳性運動失調による歩行障害が最多であり,緩徐衝動性眼球運動や眼振を伴うことが多く,進行すると筋萎縮,パーキンソニズム,認知機能障害を認めることがある3)7).深部腱反射は減弱および消失することが多いが,長期経過例では亢進を認める場合もある7).本症例は家族歴と遺伝子検査によりSCA2と診断したが,発症時に四肢運動失調や緩徐衝動性眼球運動を認めず,下位運動ニューロン障害を呈している点が特徴的であった.母方の親族に家族歴を認めたが,患者本人の母がSCDに罹患していることは確認できなかった.しかし,母は小脳萎縮を指摘されたことがあるとのことであった.母が明らかな神経症状を呈していない原因としては,ATXN2遺伝子のCAGリピート伸長は不安定で世代を経ることでリピート数が短縮する場合があることが示されており7)8),母方の祖母から母に遺伝する際にリピート数が短縮した可能性は考えられた.また,本患者のように母もSCDとしては非典型的な神経症状を有している可能性も想起されたが,当院を受診しておらず詳細は不明であった.
SCA2は病理学的に小脳入出力系だけでなく黒質,線条体,淡蒼球,青斑核,脳幹・脊髄下位運動神経前角細胞,脊髄後根神経節にも神経変性が及ぶことが報告されている9).小脳性運動失調以外の神経症状として,末梢神経障害が最も高率に認められ,その多くは感覚障害あるいは運動感覚性の障害を呈する4).一方で本症例のように下位運動ニューロン障害による運動症状のみを呈する症例が約1割存在する.通常は小脳症状の出現後に下位運動ニューロン障害が現れる3)4)10).中には末梢神経障害が前景に立つSCA2の症例が報告されている11).
SCA2の運動ニューロン障害において筋萎縮性側索硬化症(amyotrophic lateral sclerosis,以下ALSと略記)と類似した病態が示唆されている.SCA2の原因遺伝子であるATXN2遺伝子の中間リピート伸長がALSの発症リスクとなることが報告されている12)13).その機序として,ATXN2遺伝子の産物であるataxin 2とTDP-43の相互作用が考えられている.生理的にataxin 2はRNAを介してTDP-43と結合すること,ataxin 2のポリグルタミン鎖伸長によりTDP-43との結合性が増し細胞質に存在するストレス顆粒において凝集しやすくなること,ataxin 2の発現量を低下させるとTDP-43過剰発現による凝集と運動ニューロン障害が緩和されることが実験的に示されている12)14).Ataxin 2とTDP-43はいずれもmRNAの安定化と翻訳調節を担うRNA結合タンパク質であり,通常は核にも局在する.ストレス顆粒において凝集することで,本来の核への局在とRNAへの作用が失われ,神経細胞の機能不全をもたらすことが考えられている.剖検例の検討で,SCA2の運動ニューロンにおいてTDP-43の核外凝集を認めることが報告されている12).ATXN2遺伝子のCAGリピート伸長により運動ニューロンでTDP-43の凝集や機能異常が起こり,ALSに類似した症状を呈する可能性が想起される.ATXN2遺伝子のCAGリピート数は通常23回以下であり,中間リピート伸長にあたる27~33回でALS発症のリスクに,病的リピート伸長にあたる34回以上ではSCA2発症の原因となるとされる13)15).しかし,中間リピート伸長でSCA2とALSの両疾患が併存する症例16),病的リピート伸長の家系でSCA2とALSが共に見られることも報告されている15).さらに,ATXN2遺伝子の中間リピート伸長を伴うALSで小脳虫部のプルキンエ細胞が減少していたという報告がある17).これらの報告からは,リピート数だけではSCA2とALSを明確に区別できない可能性が考えられる.本症例は病的リピート伸長を示しながら下位運動ニューロン障害が前景に立っており,SCA2としては初期に典型的な症状を欠いていた.“ATXN2遺伝子のCAGリピート数異常症” という枠組みでSCA2とALSの病態が重複する可能性が示唆された.下位運動ニューロン障害の鑑別疾患の一つとしてSCA2を考慮する必要があると考えられた.
Notes
本報告の要旨は,第122回日本神経学会近畿地方会で発表し,会長推薦演題に選ばれた.
文献
- 1) Pulst SM, Nechiporuk A, Nechiporuk T, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 1996;14:269-276.
- 2) Sanpei K, Takano H, Igarashi S, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 1996;14:277-284.
- 3) Stezin A, Venkatesh SD, Thennarasu K, et al. Non-ataxic manifestations of Spinocerebellar ataxia-2, their determinants and predictors. J Neurol Sci 2018;394:14-18.
- 4) Linnemann C, Tezenas du Montcel S, Rakowicz M, et al. Peripheral neuropathy in spinocerebellar ataxia type 1, 2, 3, and 6. Cerebellum 2016;15:165-173.
- 5) Eisen A, Kuwabara S. The split hand syndrome in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2012;83:399-403.
- 6) Jacobi H, du Montcel ST, Bauer P, et al. Long-term disease progression in spinocerebellar ataxia types 1, 2, 3, and 6: a longitudinal cohort study. Lancet Neurol 2015;14:1101-1108.
- 7) Cancel G, Dürr A, Didierjean O, et al. Molecular and clinical correlations in spinocerebellar ataxia 2: a study of 32 families. Hum Mol Genet 1997;6:709-715.
- 8) Laffita-Mesa JM, Paucar M, Svenningsson P. Ataxin-2 gene: a powerful modulator of neurological disorders. Curr Opin Neurol 2021;34:578-588.
- 9) Estrada R, Galarraga J, Orozco G, et al. Spinocerebellar ataxia 2 (SCA2): morphometric analyses in 11 autopsies. Acta Neuropathol 1999;97:306-310.
- 10) Escorcio Bezerra ML, Pedroso JL, Braga-Neto P, et al. Pattern of peripheral nerve involvement in spinocerebellar ataxia type 2: a neurophysiological assessment. Cerebellum 2016;15:767-773.
- 11) Inada R, Hirano M, Oka N, et al. Phenotypic and molecular diversities of spinocerebellar ataxia type 2 in Japan. J Neurol 2021;268:2933-2942.
- 12) Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010;466:1069-1075.
- 13) Sproviero W, Shatunov A, Stahl D, et al. ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol Aging 2017;51:178.e1-e9.
- 14) Becker LA, Huang B, Bieri G, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017;544:367-371.
- 15) Tazen S, Figueroa K, Kwan JY, et al. Amyotrophic lateral sclerosis and spinocerebellar ataxia type 2 in a family with full CAG repeat expansions of ATXN2. JAMA Neurol 2013;70:1302-1304.
- 16) Nezhad HG, Franklin JP, Alix JJ, et al. Simultaneous ALS and SCA2 associated with an intermediate-length ATXN2 CAG-repeat expansion. Amyotroph Lateral Scler Frontotemporal Degener 2021;22:579-582.
- 17) Tan RH, Kril JJ, McGinley C, et al. Cerebellar neuronal loss in amyotrophic lateral sclerosis cases with ATXN2 intermediate repeat expansions. Ann Neurol 2016;79:295-305.