要旨
組織球症(histiocytosis,以下HCと略記)は単球系細胞が様々な臓器に集簇し傷害する炎症性骨髄腫瘍で,ランゲルハンス細胞組織球症(Langerhans cell histiocytosis,以下LCHと略記)やエルドハイム-チェスター病(Erdheim–Chester disease,以下ECDと略記)が含まれ,BRAFV600Eを代表とする分裂促進因子活性化タンパク質キナーゼ(mitogen-activated protein kinase,以下MAPKと略記)経路の活性化遺伝子変異を認める.LCH例の数%は,診断後数年以上経過し初期症状が消失した時期に,左右対称性の小脳病変や脳萎縮が現れ小脳失調や高次脳機能障害が生じる.ECDにも同様の中枢神経変性症(neurodegeneration,以下NDと略記)がある.近年,MAPK阻害剤によりこれらが改善することが報告された.この中枢神経障害を原因不明のNDやHC例から早期に見出しMAPK阻害剤により治療すれば,改善が期待できる.
Abstract
Histiocytoses, including Langerhans cell histiocytosis (LCH) and Erdheim–Chester disease (ECD), are inflammatory myeloid tumors in which monocyte lineage cells aggregate in various organs, causing tissue damage. Most of these tumors harbor oncogenic mutations in mitogen-activated protein kinase (MAPK) pathway genes, typified by BRAFV600E. Some patients with LCH develop bilateral symmetrical cerebellar lesions and brain atrophy several years after diagnosis when the initial symptoms disappear, leading to cerebellar ataxia and higher cerebral dysfunction. A similar neurological disorder has also been reported in ECD. This neurological disorder can be improved with MAPK inhibitors. When patients with this neurological disorder are identified among neurodegeneration of unknown etiology or histiocytosis patients and treated early with MAPK inhibitors, the disorder can be reversible.
はじめに
組織球症(histiocytosis,以下HCと略記)は,単球系細胞が様々な臓器に集簇し傷害する炎症性骨髄腫瘍の総称である.分裂促進因子活性化タンパク質キナーゼ(mitogen-activated protein kinase,以下MAPKと略記)経路は,細胞膜上の成長因子などの受容体から,細胞質内のRAS→RAF(ARAF/BRAF)→MEK→ERKへ連なる,核内へ転写シグナルを伝える経路で,細胞の分化や増殖などに重要な役割があり,がんの約40%はこの経路の遺伝子に活性化変異を認める1).多くのHCの病的細胞にもBRAFV600Eを代表とするMAPK経路の活性化遺伝子変異を認める.最も多いHCは小児に好発するランゲルハンス細胞組織球症(Langerhans cell histiocytosis,以下LCHと略記)であるが,発症から数年以上経過し初期病変が消失した時期に小脳失調や高次脳機能障害が非可逆的に進行する中枢神経変性症(neurodegeneration,以下NDと略記)を続発することが30年前から知られていた2).また,成人に多いエルドハイム-チェスター病(Erdheim–Chester disease,以下ECDと略記)でも診断時から徐々に進行する同様のNDの存在がわかってきた3).近年,これらがMAPK阻害剤によって改善する可能性が報告されている4).しかし,このHC関連中枢神経変性症(HCND)は極めて認知度が低いため,原因不明のNDやHC患者の中にHCNDが埋もれている可能性がある.本総説では,HCNDに関する知見をまとめ,スクリーニングの方策と治療について述べる.
HCの疫学と臨床像
HCNDを続発する代表的なHCにはLCH5)とECD6)がある.両者の臨床像をTable 1にまとめる.
Table 1 Clinical characteristics of Langerhans cell histiocytosis and Erdheim–Chester disease.
|
|
Langerhans cell histiocytosis |
Erdheim–Chester disease |
| Susceptible age |
Infants (<3 years old) |
Adults (around 50 years old) |
| Gender |
Male predominant in infants |
Male predominant |
| Incidence in Japan |
About 100 cases/year |
Several cases/year |
| Pathologic cells |
Positive for CD1a and CD207
Immature dendritic cell* |
Positive for CD68 and negative
for CD1a foamy macrophage** |
Involved
organ |
Bone |
80–90% (osteolytic and common
in cranial bone) |
80% (osteosclerotic and common
in bilateral lower extremity) |
| Skin/Mucosa |
40–50% |
25% |
| Xanthoma |
None |
30% |
| Lymph node |
20% |
None |
| Hematopoietic |
15% |
10% |
| Liver |
15% |
<10% |
| Spleen |
10% |
<10% |
| Lung |
10% |
30% |
| Thymus |
10% |
None |
| CDI |
10% |
40% |
| APHD |
5% |
40% |
| Peri-aorta*** |
None |
50% |
| Peri-renal*** |
None |
70% |
| Spontaneous regression |
Sometimes in solitary bone
or isolated skin lesions |
None |
*Coffee bean like twisted cell, **Presence of multinucleated giant cells with a central ring of nuclei (Touton-type giant cells), ***Fibrotic lesions. CDI, central diabetes insipidus; APHD, anterior pituitary hormone deficiency.
LCHは乳幼児に好発するが,日本での発症数は年間約100例で,その20~30%は成人発症と推計される.小児では男児にやや多いが,成人では男女差はない.診断は病変組織の病理所見によりなされ,病的組織球はCD1a+/CD207+の未熟樹状細胞でコーヒー豆様のくびれた核が特徴である.溶骨性病変が最多で,小児では頭蓋骨に多く難治性中耳炎や頭蓋腫瘤を呈することがある.その他,皮膚/粘膜や造血器・肝臓・脾臓・肺・下垂体病変等がみられる.孤発性の骨病変や皮膚単独病変の場合には自然治癒することがある.
ECDはほとんどが成人で男性に多く,日本での発症数は年間数例と推計される.診断時,20%近くの例にLCHの既往または併存がある.病変組織の病理所見により診断され,病的組織球はCD68+/CD1a−の泡沫状マクロファージで,核がリング状に配列された多核のTouton型巨細胞の出現が特徴である.硬化性骨病変が最多で,両側性で下肢に多い.その他,皮膚/粘膜や造血器・肝臓・脾臓・肺・下垂体等病変がみられるが,大血管や腎周囲の線維化病変が特徴である.自然治癒することはない.
このほか,比較的頻度の高いHCとして,乳幼児に好発する若年性黄色肉芽腫(JXG)(日本での発症数50例/年)や若年成人に好発するRosai-Dorfman病(RDD)(日本での発症数<5例/年)がある.頭蓋内腫瘤性病変をJXG・RDDの2~5%に認め7)8),このような例にNDのリスク因子となるBRAFV600E変異の報告されている9)10)ことから,JXGやRDDに続発するNDが存在する可能性はあるが,現在のところ報告は見当たらない.
LCHおよびECDにおけるMAPK経路の遺伝子変異
2010年にLCHの約半数で病変組織にBRAFV600E変異が検出されることが報告され11),これを契機にMAP2K1などMAPK経路の遺伝子に活性化変異が次々に同定された.90%のLCHはMAPK経路に活性化遺伝子変異があり12),この変異はドライバー変異である13).ECDも半数近くにBRAFV600E変異があり14),NRASやKRAS等を含め90%にMAPK経路に活性化遺伝子変異が検出される12).
HCNDの疫学
LCH関連ND(LCHND)では,脳MRI異常がまず現れ(放射線学的ND[rND]),その後,神経症状が出現(臨床的ND[cND])する.成人LCH例がLCHNDを続発することは極めてまれで15),LCHNDのほとんどは小児LCHに続発している.小児LCHの24%がrNDを続発する16).日本の小児LCHの発症数は年間70例ほどであり,LCHrND新規例は年間15例ほどと推計される.日本の小児多発骨型/多臓器型LCHのコホート17)18)とフランスのコホート19)では,LCH診断後15年の時点のLCHcNDの累積続発率は6%近くであることから,日本でのLCHcND新規例は年間4例ほどと推計される.LCHcNDと診断された年齢の中央値は,日本のコホートでは7.9歳(幅4.0~14.5),フランスのコホートでは9.2歳(幅3.4~25.8)である.続発率に男女差はない.
ECDの38%に中枢神経症状を認め,その半数,すなわち20%近くがND様の画像所見を認める20).日本でのECDの発症数は年間数例であり,ECD関連cND(ECDcND)新規例は年間1例ほどと推計される.ECDNDの報告例はすべて50歳前後の成人で,ほとんどが男性である21)22).
HCNDの病態
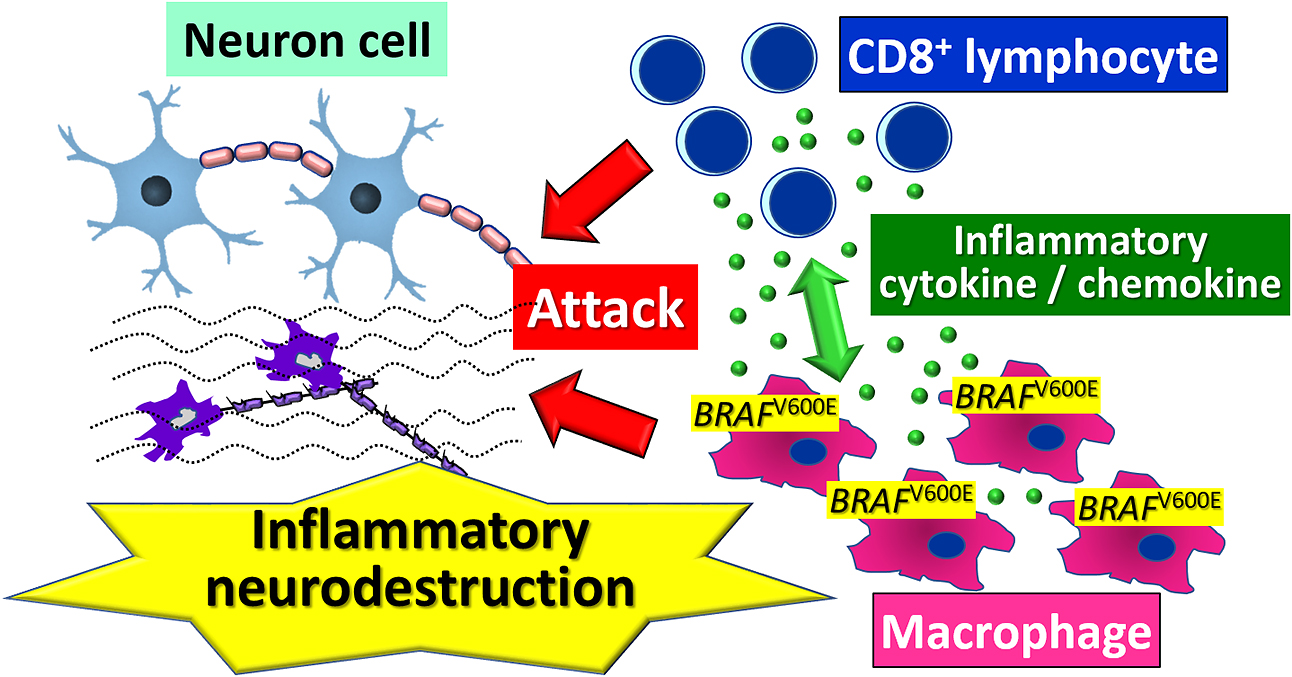
LCHNDの脳病変には,CD1a+LCH細胞を認めずCD8+Tリンパ球とマクロファージの浸潤がある23).LCHNDのほとんどはBRAFV600E変異陽性で,LCHND続発例は活動性のLCH病変がなくても末梢血や髄液中にBRAFV600E変異が検出される例があり,脳病変部の血管周囲に脳常在マクロファージ(ミクログリア)のマーカー陰性でBRAFV600E変異陽性の組織球が浸潤し,病変部位では炎症性サイトカイン/ケモカインであるオステオポンチン(OPN)が高発現している4).LCH細胞の発現プロファイルは卵黄囊造血由来の皮膚常在マクロファージ(ランゲルハンス細胞)ではなく骨髄由来の未熟骨髄樹状細胞に類似すること24)から,LCHNDの原因はLCH細胞と起源を同じくする骨髄由来前駆細胞の脳血管周囲への浸潤による炎症性神経破壊と推論されている(Fig. 1).
ECDNDの脳には,リンパ球浸潤とグリオーシス・神経細胞の喪失を認め21),泡沫状マクロファージの浸潤を認める例があり22),ECDNDも炎症性神経破壊が原因と考えられる.ECDND例もほとんどがBRAFV600E変異陽性である21).
HCNDの臨床像と経過
LCH診断時にLCHNDを認めることは極めてまれで,多くの場合,LCH診断後3年ほどを経て初期の骨病変等が消失した時期に,MRIで小脳歯状核にT2高信号,基底核にT1高信号の左右対称性の造影効果のない異常所見が出現する(LCHrND)25).橋被蓋,錐体路を含む橋や小脳脚,大脳白質内にもしばしば異常信号が出現し,小脳白質や松果体に囊胞病変がみられる例がある26)27).これらの画像所見はほとんどが徐々に悪化し,小脳や大脳の萎縮が現れる26)27)(Fig. 2).MRI異常の出現時,半数の例でLCHの活動性病変は消失している25).MRI異常のある例の25%は,数年以内に小脳失調などの運動障害や,知能低下や学習障害・性格変化などの高次脳機能障害が出現する(LCHcND)19)25)(Fig. 3).神経症状の進行は個人差が大きく,軽度の運動失調のみで経過する例から数年で植物状態に至る例まである28).LCHcNDの累積続発率は,5年で1~2%,10年で4%,15年で6%17)~19)と,LCH診断から5年以降に増加する.日本のコホートでは,LCH診断からの観察期間の中央値12年で,317例中15例にLCHcNDを認め,このうち10例(67%)は中枢性尿崩症(central diabetes insipidus,以下CDIと略記)を,6例(40%)は下垂体前葉ホルモン分泌不全(anterior pituitary hormone deficiency,以下APHDと略記)を伴い,計11例(73%)がいずれかを伴っていた.全例において,これらCDI・APHDは,LCHcNDに先行または同時に発症し,LCHNDが先行した例はなかった.神経症状の出現時にLCHの活動性病変を認めたのは4例(27%)のみであった.

ECDNDで脳MRI異常や神経症状が確認される時期は,ECD診断に先行または同時が60%近く,ECD診断後2年以内が30%近くを占める.80%は神経症状出現時に骨病変や後腹膜線維症などのECDの活動性病変を認める.半数以上は頭蓋顔面骨の病変を伴い,30%近くにCDIを認める.初発症状は歩行障害が多く,小脳症状や行動・認知障害等が徐々に進行し,30%近くが2~4年で死亡する21)22).脳MRIでLCHNDと同様の所見を認める.造影効果のある結節性病変を合併する例は神経症状の進行が早い22).半数の例は,頸・胸・腰部脊髄にもT2高信号の造影効果のない斑状病変を認める.
HD-ND続発のリスク因子
眼窩や上顎などの顔面骨,側頭骨,頭蓋底,副鼻腔を構成する骨に病変のあるLCH症例はCDIの併発リスクが高い(2.6倍)ため,これらは「CNSリスク部位」と呼ばれるが29),LCHNDのリスク因子でもある19).CDI併発した例を5年間以上観察すると76%がLCHrNDを続発する30).LCHcND続発率は,CDI併発例(13.2~20.0% vs. 0.7~1.9%)18)19),再発例(特にCNS部位への)18)19)に有意に高い.フランスのコホートでもLCHcNDのほとんど(94%)はLCH病変組織でBRAFV600E変異を認め,LCHcNDの累積続発率はBRAFV600E変異陽性群で有意に高い(22.8% vs. 1.3%)19).
ECDNDのリスク因子は解析されていないが,ECDcNDの例には,頭蓋顔面骨の病変21)22),CDI併発21),BRAFV600E変異が多い21)ことから,LCHNDと同様に,これらはECDNDのリスク因子と考えられる.
HCNDのバイオマーカー
神経炎症性疾患である多発性硬化症31)32)と同様に,LCHNDにおいても炎症性サイトカインのインターロイキン(IL)-17やOPN,神経軸索の細胞骨格成分であるニューロフィラメント軽鎖(neurofilament light chain,以下NFLと略記)がバイオマーカーとなる.IL-17はLCHの病態形成に強く関わっている33).血漿および髄液中IL-17はLCHNDで上昇し34),LCHNDの診断に有用なマーカーとなる.OPNもLCHの病態形成に強く関わり35)36),血漿および髄液中OPNはCDIを併発したLCHで上昇する37).OPNは,LCHNDの脳病変で高発現し,LCHcNDの髄液中で上昇し4),LCHNDの診断に有用なマーカーとなる.髄液38)および血漿中39)NFLはLCHNDを続発したLCHで上昇する.また,髄液中のNFL値は,後述するMAPK阻害剤による治療に反応して低下し40),LCHNDの診断,および,治療反応性の評価に有用となる.
LCHが寛解状態にもかかわらずLCHrND例においてBRAFV600E変異が末梢血単核球中に3/5例(60%)で検出されたとの報告41),LCHが寛解状態にもかかわらずLCHcND例においてBRAFV600E変異が末梢血単核球中に8/36例(22%)・髄液中に2/20例(10%)で検出されたとの報告4)がある.よってLCHが寛解状態にもかかわらず末梢血単核球や髄液中にBRAFV600E変異が検出された例はLCHNDを疑うべきである.
ECD42)とLCH43)でサイトカイン/ケモカインのパターンは多少異なるので,LCHと同様にIL-17やOPNがECDNDのバイオマーカーとなるかは不明である.炎症性神経破壊という共通した病態からすると,NFLはECDNDにおいてもバイオマーカーとなると考えられる.ECDND例の多くは,ECDの活動性病変を伴っているため21)22),末梢血中にBRAFV600E変異が検出される例が多い21).
HCNDの治療
LCHNDに対して免疫グロブリン(IVIG)療法やシタラビン(Ara-C)療法が,ECDNDに対してインターフェロン(IFN)-α療法が試みられてきたが,再現性のある有効性が明らかな治療法はなかった.IVIG療法は神経症状の進行を遅らせる可能性はあるが改善させることはない28)44).Ara-C療法によりLCHcNDが改善したとの報告45)があるが,LCHの初期治療としてAra-C療法46)を用いても数%はLCHNDを続発すること17)18),LCHNDに対してAra-C療法の有効性を検証した報告が他にないことから,その効果は不明である.IFN-α療法は中枢神経病変のあるECDの生存率を向上させるが47),ECDNDを改善させない21).
LCH48)およびECD49)に対するMAPK阻害剤の有効性は確立している.日本では2023年9月時点でいずれのMAPK阻害剤もHCに対して保険承認されていないが,BRAFV600E変異のあるECDにBRAF阻害剤であるベムラフェニブ(VMF)50)が,遺伝子変異にかかわらず成人HCにMEK阻害剤であるコビメチニブ51)がFDAにより承認されている.最近,MAPK阻害剤によりHCNDが改善したという報告が蓄積されつつある.
McClainらは4),4例のLCHcNDにBRAF阻害剤を投与した.神経症状の出現直後の1例ではMRI所見の改善と症状の消失,神経症状出現後1~4年の2例ではMRI所見と症状の軽度改善を得たが,神経症状出現後10年以上経過した例ではMRI所見・症状ともに改善は得られなかった.Ecksteinらは52),13例のLCHND(LCHrND 4例,LCHcND 9例)にBRAF阻害剤またはMEK阻害剤を投与した.LCHrNDの4例全てにMRI所見の改善,LCHcNDの9例(神経症状出現後の中央値23か月[幅0か月~12年])中8例に神経症状の改善を得たが,1例は不変だった.不変だった1例は神経症状の出現後9年経過した例であった.これらのことからするとLCHNDの早期,すなわち,LCHrNDの段階あるはLCHcNDとなって数年以内にMAPK阻害剤で治療すると,改善が期待できる.しかし,いずれの報告でも,治療中断により増悪し,治療期間について今後検討が必要である.ECDNDに対するMAPK阻害剤の報告は少ないが,Chiappariniら21)は,IFN-α不応のECDcNDの2例にVMFを投与し,MRI所見・神経症状ともに著明な改善を得た報告している.
悪性黒色腫では,BRAF阻害剤の単剤治療は耐性化を生むが53),LCHやECDにおいて耐性化は報告されていない6)54).HC例に対するVMFの副作用として,皮疹や光線過敏が半数近くにみられる55).成人では皮膚扁平上皮癌が問題となり50),早期切除の機会を逸しないように皮膚所見に注意すべきである.
HCNDのスクリーニング検査と診断基準案
HCNDはMAPK阻害剤によって改善が期待できる中枢神経障害であり,早期に治療介入したほうが治療効果は高い.よって,HCNDを早期に診断することが重要であり,そのためには,原因不明のNDやHC患者の中にHCNDがいないか,スクリーニングする必要がある.
原因不明のNDのスクリーニングとして,特徴的なMRI所見の確認,幼少時のLCH(特に頭蓋顔面骨病変)の病歴聴取,X線やPET検査による骨病変の検索,末梢血や髄液中のBRAFV600E変異アレル検索,CDI/APHDの検索,血液と髄液中IL-17やOPNの測定,が挙げられる.特徴的なMRI所見がある例において,HCの既往や活動性病変が証明できればDefinitive HCcND,HCの既往や活動性病変が明らかでなくてもBRAFV600E変異アレルの検出またはCDI/APHDの併発,IL-17/OPN高値のいずれかがあればPossible HCcND(Table 2)と診断してよいと考える.
Table 2 Proposed diagnostic criteria for histiocytosis-related neurodegeneration.
| <Major criteria> |
| 1. Presence of history* or active lesions** of histiocytosis |
| 2. Typical findings on brain MRI |
| 3. Presence of cerebellar ataxia or higher brain dysfunction |
| <Minor criteria> |
| 1. Positive for BRAFV600E mutation allele in blood or cerebrospinal fluid |
| (but only under no malignant diseases other than histiocytosis with BRAFV600E mutation) |
| 2. Presence of central diabetes insipidus or anterior pituitary hormone deficiency |
| 3. Elevation of IL-17 or osteopontin levels in blood or cerebrospinal fluid |
| (but only under no active lesions of histiocytosis) |
| <Reference findings> |
| 1. Elevation of NFL levels in blood or cerebrospinal fluid |
| (but only under no other central nervous disorders showing elevated NFL) |
| Definitive HC-cND: fulfilled all of major criteria |
| Definitive HC-rND: fulfilled the major criteria of 1 and 2 (If the MRI findings are minimum, fulfill reference findings) |
| Probable HC-cND: fulfilled the major criteria of 2 and 3, and fulfilled at least one of the minor criteria |
*especially craniofacial lesions; **especially bone lesions. NFL, neurofilament light chain; HC, histiocytosis; cND, clinical neurodegeneration; rND, radiological neurodegeneration.
HCのスクリーニングとして,頭蓋顔面骨病変やCDI/APHDのある例や再発例の脳MRIの定期検査,小脳失調などの神経所見の検索,NFLの測定,HCが寛解状態の例における末梢血BRAFV600E変異アレル検索が挙げられる.特徴的なMRI所見に加え,神経症状があればDefinitive HCcND,神経症状がない場合はDefinitive HCrND(Table 2)として診断してよいと考える.
今後の展望
原因不明のNDおよびHC例をスクリーニングしてHCNDを早期に診断し,MAPK阻害剤による治療で重大な中枢神経障害を回避する戦略の確立が望まれる.令和5年度からAMED難治性疾患実用化研究事業「組織球症に続発する中枢神経変性症の診断・治療エビデンスの創出」研究班(代表:塩田曜子)が発足した.まず,HCNDのレジストリー構築に着手し,NFLなどの客観的指標となるバイオマーカーを用いMAPK阻害剤の臨床試験56)を進める方針である.
文献
- 1) Guo YJ, Pan WW, Liu SB, et al. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med 2020;19:1997-2007.
- 2) Grois N, Barkovich AJ, Rosenau W, et al. Central nervous system disease associated with Langerhans’ cell histiocytosis. Am J Pediatr Hematol Oncol 1993;15:245-254.
- 3) Fukazawa T, Tsukishima E, Sasaki H, et al. Erdheim-Chester disease and slowly progressive cerebellar dysfunction. J Neurol Neurosurg Psychiatry 1995;58:238-240.
- 4) McClain KL, Picarsic J, Chakraborty R, et al. CNS Langerhans cell histiocytosis: Common hematopoietic origin for LCH-associated neurodegeneration and mass lesions. Cancer 2018;124:2607-2620.
- 5) Morimoto A, Oh Y, Shioda Y, et al. Recent advances in Langerhans cell histiocytosis. Pediatr Int 2014;56:451-461.
- 6) Haroche J, Cohen-Aubart F, Amoura Z. Erdheim-Chester disease. Blood 2020;135:1311-1318.
- 7) Janssen D, Harms D. Juvenile xanthogranuloma in childhood and adolescence: a clinicopathologic study of 129 patients from the kiel pediatric tumor registry. Am J Surg Pathol 2005;29:21-28.
- 8) Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood 2018;131:2877-2890.
- 9) Picarsic J, Pysher T, Zhou H, et al. BRAF V600E mutation in Juvenile Xanthogranuloma family neoplasms of the central nervous system (CNS-JXG): a revised diagnostic algorithm to include pediatric Erdheim-Chester disease. Acta Neuropathol Commun 2019;7:168.
- 10) Cronin C, McLaughlin R, Lane L, et al. Case report: BRAF-inhibitor therapy in BRAF-mutated primary CNS tumours including one case of BRAF-mutated Rosai-Dorfman disease. Front Med (Lausanne) 2022;9:1070828.
- 11) Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010;116:1919-1923.
- 12) Durham BH, Lopez Rodrigo E, Picarsic J, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med 2019;25:1839-1842.
- 13) Berres ML, Lim KP, Peters T, et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 2014;211:669-683.
- 14) Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood 2012;120:2700-2703.
- 15) Spagnolo F, Leopizzi E, Cardamone R, et al. Neurodegeneration in the course of Langerhans cell histiocytosis. Neurol Sci 2012;33:605-607.
- 16) Laurencikas E, Gavhed D, Stalemark H, et al. Incidence and pattern of radiological central nervous system Langerhans cell histiocytosis in children: a population based study. Pediatr Blood Cancer 2011;56:250-257.
- 17) Sakamoto K, Morimoto A, Shioda Y, et al. Central diabetes insipidus in pediatric patients with Langerhans cell histiocytosis: Results from the JLSG-96/02 studies. Pediatr Blood Cancer 2019;66:e27454.
- 18) Sakamoto K, Morimoto A, Shioda Y, et al. Long-term complications in uniformly treated paediatric Langerhans histiocytosis patients disclosed by 12 years of follow-up of the JLSG-96/02 studies. Br J Haematol 2021;192:615-620.
- 19) Heritier S, Barkaoui MA, Miron J, et al. Incidence and risk factors for clinical neurodegenerative Langerhans cell histiocytosis: a longitudinal cohort study. Br J Haematol 2018;183:608-617.
- 20) Cohen Aubart F, Idbaih A, Galanaud D, et al. Central nervous system involvement in Erdheim-Chester disease: an observational cohort study. Neurology 2020;95:e2746-e2754.
- 21) Chiapparini L, Cavalli G, Langella T, et al. Adult leukoencephalopathies with prominent infratentorial involvement can be caused by Erdheim-Chester disease. J Neurol 2018;265:273-284.
- 22) Riso V, Nicoletti TF, Rossi S, et al. Neurological Erdheim-Chester disease manifesting with subacute or progressive cerebellar ataxia: novel case series and review of the literature. Brain Sci 2022;13:26.
- 23) Grois N, Prayer D, Prosch H, et al. Neuropathology of CNS disease in Langerhans cell histiocytosis. Brain 2005;128:829-838.
- 24) Allen CE, Li L, Peters TL, et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J Immunol 2010;184:4557-4567.
- 25) Wnorowski M, Prosch H, Prayer D, et al. Pattern and course of neurodegeneration in Langerhans cell histiocytosis. J Pediatr 2008;153:127-132.
- 26) Martin-Duverneuil N, Idbaih A, Hoang-Xuan K, et al. MRI features of neurodegenerative Langerhans cell histiocytosis. Eur Radiol 2006;16:2074-2082.
- 27) Prosch H, Grois N, Wnorowski M, et al. Long-term MR imaging course of neurodegenerative Langerhans cell histiocytosis. AJNR Am J Neuroradiol 2007;28:1022-1028.
- 28) Imashuku S, Fujita N, Shioda Y, et al. Follow-up of pediatric patients treated by IVIG for Langerhans cell histiocytosis (LCH)-related neurodegenerative CNS disease. Int J Hematol 2015;101:191-197.
- 29) Grois N, Potschger U, Prosch H, et al. Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatr Blood Cancer 2006;46:228-233.
- 30) Grois N, Prayer D, Prosch H, et al. Course and clinical impact of magnetic resonance imaging findings in diabetes insipidus associated with Langerhans cell histiocytosis. Pediatr Blood Cancer 2004;43:59-65.
- 31) Wen SR, Liu GJ, Feng RN, et al. Increased levels of IL-23 and osteopontin in serum and cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol 2012;244:94-96.
- 32) Sen MK, Hossain MJ, Mahns DA, Brew BJ. Validity of serum neurofilament light chain as a prognostic biomarker of disease activity in multiple sclerosis. J Neurol 2023;270:1908-1930.
- 33) Coury F, Annels N, Rivollier A, et al. Langerhans cell histiocytosis reveals a new IL-17A-dependent pathway of dendritic cell fusion. Nat Med 2008;14:81-87.
- 34) Ismail MB, Akefeldt SO, Lourda M, et al. High levels of plasma interleukin-17A are associated with severe neurological sequelae in Langerhans cell histiocytosis. Cytokine 2020;126:154877.
- 35) Oh Y, Oh I, Morimoto J, et al. Osteopontin has a crucial role in osteoclast-like multinucleated giant cell formation. J Cell Biochem 2014;115:585-595.
- 36) Oh Y, Morimoto A, Shioda Y, et al. High serum osteopontin levels in pediatric patients with high risk Langerhans cell histiocytosis. Cytokine 2014;70:194-197.
- 37) Li N, Cui L, Ma H, et al. Osteopontin is highly secreted in the cerebrospinal fluid of patient with posterior pituitary involvement in Langerhans cell histiocytosis. Int J Lab Hematol 2020;42:788-795.
- 38) Gavhed D, Akefeldt SO, Osterlundh G, et al. Biomarkers in the cerebrospinal fluid and neurodegeneration in Langerhans cell histiocytosis. Pediatr Blood Cancer 2009;53:1264-1270.
- 39) Sveijer M, von Bahr Greenwood T, Jädersten M, et al. Screening for neurodegeneration in Langerhans cell histiocytosis with neurofilament light in plasma. Br J Haematol 2022;198:721-728.
- 40) Henter JI, Kvedaraite E, Martín Muñoz D, et al. Response to mitogen-activated protein kinase inhibition of neurodegeneration in Langerhans cell histiocytosis monitored by cerebrospinal fluid neurofilament light as a biomarker: a pilot study. Br J Haematol 2022;196:248-254.
- 41) Shimizu S, Sakamoto K, Kudo K, et al. Detection of BRAF V600E mutation in radiological Langerhans cell histiocytosis-associated neurodegenerative disease using droplet digital PCR analysis. Int J Hematol. doi: 10.1007/s12185-023-03588-w. (in printing)
- 42) Arnaud L, Gorochov G, Charlotte F, et al. Systemic perturbation of cytokine and chemokine networks in Erdheim-Chester disease: a single-center series of 37 patients. Blood 2011;117:2783-2790.
- 43) Morimoto A, Oh Y, Nakamura S, et al. Inflammatory serum cytokines and chemokines increase associated with the disease extent in pediatric Langerhans cell histiocytosis. Cytokine 2017;97:73-79.
- 44) Gavhed D, Laurencikas E, Akefeldt SO, et al. Fifteen years of treatment with intravenous immunoglobulin in central nervous system Langerhans cell histiocytosis. Acta Paediatr 2011;100:e36-e39.
- 45) Allen CE, Flores R, Rauch R, et al. Neurodegenerative central nervous system Langerhans cell histiocytosis and coincident hydrocephalus treated with vincristine/cytosine arabinoside. Pediatr Blood Cancer 2010;54:416-423.
- 46) Morimoto A, Shioda Y, Imamura T, et al. Intensified and prolonged therapy comprising cytarabine, vincristine and prednisolone improves outcome in patients with multisystem Langerhans cell histiocytosis: results of the Japan Langerhans Cell Histiocytosis Study Group-02 Protocol Study. Int J Hematol 2016;104:99-109.
- 47) Arnaud L, Hervier B, Néel A, et al. CNS involvement and treatment with interferon-α are independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood 2011;117:2778-2782.
- 48) Donadieu J, Larabi IA, Tardieu M, et al. Vemurafenib for refractory multisystem Langerhans cell histiocytosis in children: an international observational study. J Clin Oncol 2019;37:2857-2865.
- 49) Haroche J, Cohen-Aubart F, Emile JF, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAF(V600E)-mutated Erdheim-Chester disease. J Clin Oncol 2015;33:411-418.
- 50) Diamond EL, Subbiah V, Lockhart AC, et al. Vemurafenib for BRAF V600-mutant Erdheim-Chester disease and Langerhans cell histiocytosis: analysis of data from the Histology-independent, Phase 2, open-label VE-BASKET study. JAMA Oncol 2018;4:384-388.
- 51) Diamond EL, Durham BH, Ulaner GA, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 2019;567:521-524.
- 52) Eckstein OS, Visser J, Rodriguez-Galindo C, et al. Clinical responses and persistent BRAF V600E(+) blood cells in children with LCH treated with MAPK pathway inhibition. Blood 2019;133:1691-1694.
- 53) Grimaldi AM, Simeone E, Festino L, et al. Novel mechanisms and therapeutic approaches in melanoma: targeting the MAPK pathway. Discov Med 2015;19:455-461.
- 54) Jouenne F, Benattia A, Tazi A. Mitogen-activating protein kinase pathway alterations in Langerhans cell histiocytosis. Curr Opin Oncol 2021;33:101-109.
- 55) Mohapatra D, Gupta AK, Haldar P, et al. Efficacy and safety of vemurafenib in Langerhans cell histiocytosis (LCH): a systematic review and meta-analysis. Pediatr Hematol Oncol 2023;40:86-97.
- 56) Shioda Y, Sakamoto K, Morimoto A. New biomarker paves the way for a clinical trial for neurodegeneration in Langerhans cell histiocytosis. Br J Haematol 2022;198:623-624.