要旨
症例は52歳男性.小児期より難聴,40歳代より下肢遠位の潰瘍や骨髄炎を繰り返した.4年前より歩行障害,1ヶ月前より構音障害が出現した.認知機能低下,両下肢近位筋の筋力低下,四肢の腱反射亢進,小脳失調,両下肢深部覚優位の感覚障害,排尿障害を認め,神経伝導検査ではSNAPが導出不能だった.祖母に同様の症状があり,DNA methyltransferase 1(DNMT1)遺伝子のp.Y495H変異を認めhereditary sensory and autonomic neuropathy 1E(HSAN1E)と診断した.HSAN1Eで腱反射亢進を呈する事がある.
Abstract
A 52-year-old man had developed hearing loss since childhood, as well as recurrent foot ulcers and osteomyelitis since his forties. He presented with gait disturbance and dysarthria that had worsened over four years and a month, respectively. Neurological exams revealed cognitive impairment, proximal weakness of the lower extremities, generalized hyperrflexia, ataxia, sensory disturbances predominant in deep sensation, urinary retention, and gait instability. On nerve conduction study, no sensory nerve action potentials were evoked in the upper and lower limbs. Since his grandmother suffered from similar symptoms, we investigated genetic analysis, which revealed a missense mutation (c.1483T>C, p.Y495H) in DNA methyltransferase 1 gene. He was subsequently diagnosed with hereditary sensory and autonomic neuropathy 1E (HSAN1E). It is important to recognize that increased deep tendon reflex can be observed in HSAN1E.
はじめに
Hereditary sensory and autonomic neuropathy(HSAN)は感覚自律神経が障害される遺伝性ニューロパチーであり1),発症年齢と遺伝形式から,成人発症で常染色体顕性遺伝であるHSAN1型と,発症が生下時または幼年期で常染色体潜性遺伝であるHSAN2-4型に分類される2).HSAN1EはDNA methyltransferase 1(DNMT1)遺伝子変異による常染色体顕性遺伝性疾患であり,難聴,感覚性ニューロパチー,認知症を3主徴とする.今回我々は当初は遺伝性痙性対麻痺(複合型)を疑ったが,最終的にHSAN1Eと診断した1例を経験した.腱反射亢進を来したHSAN1Eは稀であったため,文献的考察を加え考察する.
症例
症例:52歳,男性
主訴:歩きにくい
既往歴:高血圧症,感音性難聴,足底潰瘍,中足骨骨髄炎.
家族歴:祖母が類症.母が脊髄疾患,認知症により70歳で死去.父が心疾患により64歳で死去.
生活歴:飲酒は機会飲酒,喫煙はなし.愛媛県出身.
現病歴:小児期より難聴,40歳代より足底潰瘍を繰り返すようになり,しばしば骨髄炎に至り加療を行われていた.2010年より歩行時のふらつきを自覚し,2012年にふらつきからガスボンベを運ぶ仕事ができなくなり退職した.2013年夏頃から左下肢の脱力を自覚し,近医整形外科を受診し脊柱管狭窄症と診断されリハビリテーションを行われていた.その後も症状は徐々に進行し2013年8月に当科を紹介受診した.頭部CTにて小脳萎縮を認め精査を勧めるも,骨髄炎の治療を優先し精査ができなかった.2014年3月には構音障害が出現し,骨髄炎が軽快した2014年4月に当科に入院した.
一般理学所見:身長174 cm,体重79 kg,体温36.4°C,血圧118/77 mmHg,脈拍75/分・整,SpO2 99%(室内気).心肺聴診異常なし.両足関節以遠の腫脹および足趾変形あり.
神経学的所見:意識レベルはJapan Coma Scale I-3.高次脳機能検査ではMMSE 21点(時の見当識−2,計算−4,3単語の遅延再生−1,文章書字−1)で長期記憶障害や計算障害での失点が見られた.両側視力低下,両側難聴,構音障害を認めた.項部硬直やLhermitte徴候を認めなかった.上肢の筋トーヌスは正常で,筋力低下を認めなかった.下肢は左優位に痙縮を認め,徒手筋力検査にて近位筋4レベル,遠位筋5−レベルの筋力低下を認めた.指鼻指試験,回内回外運動はともに左優位に拙劣であった.Romberg徴候は陽性で,痙性歩行から伝い歩きであった.継ぎ足歩行はできなかった.口輪筋反射および下顎反射は陰性であった.両足の腫脹のため,診察が困難だったアキレス腱反射以外では四肢の腱反射は亢進しており,病的反射を認めなかった.Hoffmann反射は両側で陽性であった.両下肢の触覚鈍麻,振動覚消失を認めた.膀胱直腸障害を認め,勃起障害が疑われた.
検査所見:血液検査では血算,肝・腎機能,電解質は正常.HbA1c 6.9%.CRP 4.12 mg/dl.ビタミンB1,ビタミンB12,葉酸,乳酸,ピルビン酸,ACE,血清銅,フェリチン,TSH,free T4,CEA,CA19-9は基準範囲内.抗核抗体,抗ds-DNA抗体,抗SS-A抗体,抗SS-B抗体,抗TPO抗体,抗HTLV-1抗体,MPO-ANCA,PR3-ANCAは全て陰性.その他,可溶性IL-2受容体1,421 U/ml(基準範囲122~496 U/ml).髄液検査で初圧160 mmH2O,細胞数0/μl,蛋白量22 mg/dl,細胞診で異型細胞を認めなかった.胸腹部造影CTでは腫瘍性病変,リンパ節腫脹は無かった.頭部MRIでは大脳皮質や小脳半球の萎縮に加えて,脳梁の菲薄化が認められた.拡散強調画像では異常信号は無かった.全脊椎MRIでは軽度の頸髄症を認めたものの,異常信号を認めなかった(Fig. 1).99mTc-ethyl cysteinate dimer single photon emission computed tomography(99mTc-ECD SPECT)では小脳を含めて血流低下部位は無かった.12誘導心電図ではHR 58 bpm,正常洞調律・整,正常軸.CVR-R 4.69%.運動神経伝導検査では右正中神経,尺骨神経ともに軽度伝導速度遅延を認め,右脛骨神経では膝窩刺激で複合筋活動電位を誘発できなかった.右尺骨神経のF波の出現頻度が軽度低下していた.右上下肢で感覚神経活動電位を誘発できなかった(Table 1).眼科では両側白内障を指摘され,聴力検査では左優位に高音漸傾型の感音性難聴を認めた.

Table 1 Nerve conduction study of the right upper and lower extremities.
|
Motor conduction study |
F wave |
Sensory conduction study |
Distal latency
(msec) |
Amplitude
(mV) |
MCV
(m/s) |
Frequency
(%) |
Minimum latency
(msec) |
Amplitude
(μV) |
SCV
(m/s) |
| Rt. median |
4.1 |
9.1 |
46.9 |
81 |
24.0 |
NE |
NE |
| Rt. Ulnar |
3.4 |
10.1 |
45.4 |
63 |
24.2 |
NE |
NE |
| Rt. Tibial |
5.3 |
8.0 |
NE* |
100 |
46.9 |
|
|
| Rt. sural |
|
|
|
|
|
NE |
NE |
Abbreviations: MCV; motor conduction velocity, NE; not evoked, CMAP; compound muscle action potential.
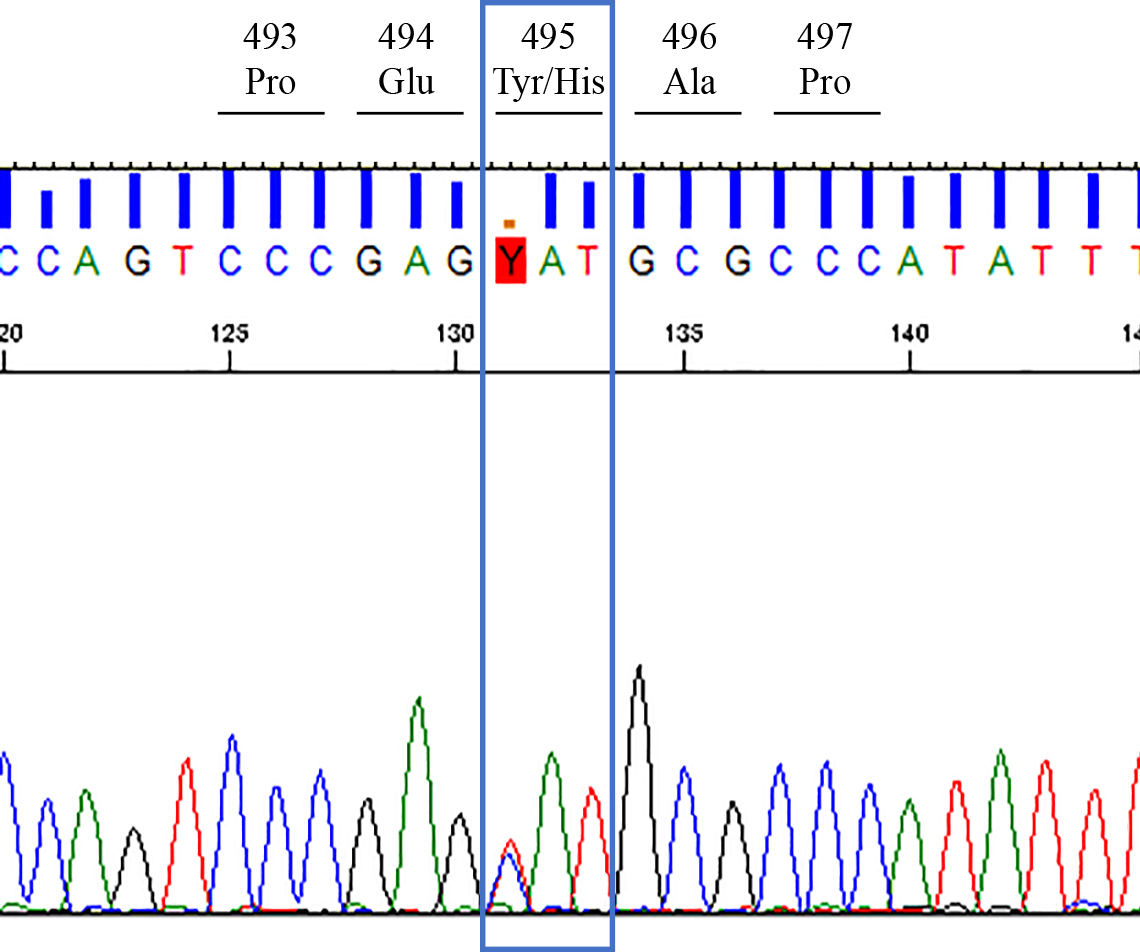
錐体路徴候,小脳性運動失調,感覚優位の末梢神経障害,家族歴,脳梁の菲薄化からは遺伝性痙性対麻痺(複合型)を鑑別に挙げ,山梨大学医学部附属病院神経内科Japan Spastic Paraplegia Research Consortium(JASPAC)へ依頼した.退院後は当科で経過観察する方針であったが,足の感染を契機に他院に入退院を繰り返しており受診が途切れていた.他院診療記録からは徐々に歩行障害,認知機能障害は進行し,2017年に胃瘻を造設し同年に死亡した.Exome解析により変異解析を行い,SPG遺伝子,alsin遺伝子,SACS遺伝子,ABCD1遺伝子に加えて,遺伝性ポリニューロパチーの関連遺伝子について網羅的に解析を行ったところ,DNMT1遺伝子でexon 20にヘテロ接合性の可能性のあるNM_001379.4:c.1483T>C(p.Tyr495His)が判明した.加えてサンガーシーケンスで同様の変異を確認でき,HSAN1Eと確定診断した(Fig. 2).当該variantはdbSNP(https://www.ncbi.nlm.nih.gov)ではrs199473692に該当した.また他院の再精査では髄液総Tau蛋白,14-3-3蛋白は陰性で,プリオン蛋白遺伝子はcodon129でMet/Met,codon219でGlu/Gluであった.SCD遺伝子解析(SCA1, 2, 3, 6, 7, 8, 12, 31, DRPLA)は陰性であった.
考察
HSANは小径有髄神経や無髄神経線維の障害により感覚神経を主体に障害され,温痛覚障害による外傷や熱傷を繰り返し,無痛性潰瘍や足趾の変形を伴う疾患である1).しかしHSAN1Eの腓腹神経では有髄線維の完全の脱落と無髄線維の減少を認めた報告例もあり,病型によっては深部覚優位の感覚障害を来す場合もある2).HSAN1EはDNMT1遺伝子関連疾患であり,DNAメチル化酵素であるDNMT1は遺伝子発現調整,クロマチン安定化に必須であるとされている3).DNMT1遺伝子関連疾患にはexon 20に変異が集中するHSAN1Eの他に,小脳性運動失調,難聴,ナルコレプシーを呈しexon 21に変異が集中するautosomal dominant cerebellar ataxia,deafness and narcolepsy(ADCA-DN)が知られている.両者はオーバーラップする事もあり,「DNMT1 methylopathy」という概念もある4).HSAN1Eは非常に稀であり,本邦ではKleinら5),Yuanら6),および渡部ら7)の報告でDNMT1遺伝子変異を確認できた.そこで,これまでDNMT1遺伝子変異を伴うHSAN1Eの臨床像が記載された報告を渉猟したところ,本例の他に23家系79例が確認できた(Table 2)4)~13).患者は39/79例が男性,40/79例が女性で性差はなく,診察時の平均年齢は22歳から65歳だった.1家系は表在覚優位の感覚障害を呈していたが,4家系は深部覚優位であり既報告通りであった2).本例の他に2例で自律神経障害,11例で小脳性運動失調を確認できた.腱反射は1家系で保たれていたが6家系では消失しており,本例のように腱反射亢進を認めた症例は無かった.HSAN1Eの発症に関わるhot spotとされているp.Tyr495に関わるDNMT1遺伝子変異を10家系で確認でき,本例と同じp.Tyr495Hisの変異に関しては3家系7)~9)を認めた.本例のように典型的に難聴や感覚性ニューロパチーを初発としたのは2家系8)9)であり,本邦での1家系7)は前頭葉徴候を初発とし,垂直性眼球運動障害,筋強剛,頭部MRIでの中脳被蓋の萎縮所見から当初は進行性核上性麻痺が疑われていた.DNMT1遺伝子変異では末梢神経障害だけでなく中枢神経障害を起こす事もあり,DNMT1遺伝子解析でc.2053G>A(p.Ala685Thr)ミスセンス変異を認め痙性対麻痺を来した報告もある14).DNMT1遺伝子解析は施行されていないものの,腱反射減弱を認めHSAN1Eと臨床診断された剖検症例は報告されている15)16).病理学的所見では肉眼的に前頭葉優位のびまん性脳萎縮を認め,顕微鏡的には大脳皮質や海馬,大脳基底核,小脳に神経細胞の脱落や風船様神経細胞,軸索変性,線維性アストロサイトの増生,ミクログリアの活性化の所見が報告されている.そのうち線維性アストロサイトの増生,ミクログリアの活性化については前頭葉皮質優位であり,錐体路障害と本遺伝子変異の関連が示唆される.Baetsらの報告ではHSAN1Eの初発症状は難聴が36%と最多であり9),本例も小児期からの難聴が指摘されていた.難聴を初発として感覚性ニューロパチーを来した場合は,HSAN1Eを鑑別に挙げるべきである.
Table 2 Clinical features of hereditary sensory and autonomic neuropathy 1E (HSAN1E) patients with DNA methyltransferase 1 (
DNMT1) mutation on published data.
| Author |
Patients |
Sex |
DNMT1 mutation |
Mutation location
(exon) |
Mean age
at evaluation |
Hearing loss |
Sensory
disturbance |
Dementia |
Autonomic
dysfunction |
Deep tendon
reflex |
Cerebellar
ataxia |
| Klein5) |
11 |
6F |
p.Tyr495Cys |
20 |
38 |
11/11 |
11/11 (S, D) |
11/11 |
N/A |
N/A |
0/11 |
| Klein5) |
3 |
2F |
p.Asp490Glu-Pro491Tyr |
20 |
36 |
3/3 |
3/3 (S, D) |
3/3 |
N/A |
N/A |
2/3 |
| Klein5) |
3 |
2F |
p.Tyr495Cys |
20 |
42 |
3/3 |
3/3 (S, D) |
3/3 |
N/A |
N/A |
2/3 |
| Klein5) |
1 |
F |
p.Tyr495Cys |
20 |
40s |
1/1 |
1/1 (S, D) |
1/1 |
N/A |
N/A |
0/1 |
| Klein8) |
3 |
1F |
p.Tyr495His |
20 |
53 |
3/3 |
1/1 (D) |
1/1 |
N/A |
N/A |
N/A |
| Klein8) |
3 |
3F |
p.Tyr495Cys |
20 |
47 (1 patient) |
3/3 |
2/2 (S) |
3/3 |
N/A |
N/A |
N/A |
| Yuan6) |
1 |
M |
p.His569Arg |
21 |
41 |
1/1 |
1/1 (S<D) |
1/1 |
N/A |
− |
0/1 |
| Moghadam4) |
2 |
1F |
p.Pro507Arg |
20 |
35 |
2/2 |
2/2 (S, D) |
0/2 |
0/2 |
− |
1/2 |
| Moghadam4) |
1 |
M |
p.Lys521del |
20 |
59 |
1/1 |
1/1 (S, D) |
1/1 |
1/1 |
− |
1/1 |
| Baets9) |
1 |
F |
p.Thr481Pro |
N/A |
34 |
1/1 |
1/1 (S, D) |
1/1 |
N/A |
N/A |
N/A |
| Baets9) |
1 |
M |
p.Pro491Leu |
N/A |
48 |
1/1 |
1/1 (S, D) |
1/1 |
N/A |
N/A |
N/A |
| Baets9) |
2 |
2M |
p.Tyr524Asp |
N/A |
45 |
2/2 |
2/2 (S, D) |
0/1 |
N/A |
N/A |
N/A |
| Baets9) |
2 |
1F |
p.Ile531Asn |
N/A |
42 |
2/2 |
2/2 (S, D) |
2/2 |
N/A |
N/A |
N/A |
| Baets9) |
1 |
M |
p.Tyr495Cys |
20 |
45 |
1/1 |
1/1 (S, D) |
1/1 |
N/A |
− |
N/A |
| Baets9) |
7 |
3F |
p.Cys353Phe |
N/A |
64 |
6/7 |
2/4 (S, D) |
1/4 |
N/A |
N/A |
N/A |
| Baets9) |
10 |
9F |
p.Tyr495Cys |
20 |
50s |
10/10 |
7/7 (S, D) |
9/10 |
N/A |
N/A |
N/A |
| Baets9) |
10 |
2F |
p.Tyr495His |
20 |
50s |
8/8 |
8/8 (S, D) |
6/6 |
N/A |
N/A |
N/A |
| Baets9) |
11 |
5F |
p.Tyr495Cys |
20 |
48 |
11/11 |
11/11 (S, D) |
11/11 |
N/A |
N/A |
N/A |
| Watanabe7) |
1 |
M |
p.Tyr495His |
20 |
49 |
1/1 |
1/1 (S<D) |
1/1 |
0/1 |
− |
1/1 |
| Zheng10) |
1 |
F |
p.Tyr540Asn |
20 |
38 |
1/1 |
N/A |
1/1 |
N/A |
N/A |
1/1 |
| Catania11) |
2 |
1F |
p.Arg598Trp |
21 |
65 (1 patient) |
2/2 |
2/2 (D) |
2/2 |
1/1 |
N/A |
2/2 |
| Coelho12) |
1 |
M |
p.Leu567Pro |
21 |
22 |
1/1 |
1/1 (S<D) |
0/1 |
0/1 |
+ |
1/1 |
| Parissis13) |
1 |
F |
p.Leu566His |
21 |
33 |
1/1 |
1/1 (S, D) |
1/1 |
0/1 |
− |
0/1 |
| Our case |
1 |
M |
p.Tyr495His |
20 |
52 |
1/1 |
1/1 (S<D) |
1/1 |
1/1 |
++ |
1/1 |
Abbreviations: M; male, F; female, D; deep sensation, S; superficial sensation, N/A; not available.
血液検査では可溶性IL-2受容体 1,421 U/mlと著しく上昇していた.本例では発熱,皮膚所見,血球減少,リンパ節腫大,MRIでの異常所見を認めなかった事を考慮すると,悪性リンパ腫の可能性は低いと考える.入院中も両足の腫脹や疼痛を認め,CRP高値である事から骨髄炎が影響を及ぼしていた可能性を否定できない.
本例では神経学的に小脳性運動失調を認め,MRIで小脳萎縮があったにも関わらず,99mTc-ECD SPECTで血流低下を認めなかった原因は明らかでない.しかし小脳半球のみならず,びまん性に大脳皮質の萎縮を認めた事から血流低下がめだたなかった可能性を推測する.
また当院では運動誘発電位の施行ができなかったため客観的な根拠には欠け,足趾変形により病的反射の評価が困難だったかもしれないが,腱反射亢進および下肢痙縮からは本例における錐体路障害が示唆される.MRIでは軽度の頸髄症はあったものの,脊髄に異常信号を認めていない点からは原因とは考えにくくDNMT1遺伝子変異の様々な表現型が示唆されている.渡部らの報告も愛媛県での報告で同じp.Tyr495Hisの変異を認めており7),本例との関連は明らかではないものの,HSAN1Eが愛媛県に集積している可能性がある.今後の更なる症例蓄積による検討が期待される.
DNMT1遺伝子解析でc.1483T>C(p.Y495H)ミスセンスを有するHSAN1Eの1例を報告した.同疾患で腱反射亢進を呈する事があり,注意が必要である.
利益相反
著者全員に本論文に関連し,開示すべきCOI状態にある企業,組織,団体はいずれも有りません.
謝辞
本症例の診断・治療にあたりご尽力頂きました東北大学大学院医学科病態神経学の北本哲之先生,長崎大学医歯薬学総合研究科感染分子学教室の佐藤克也先生,鹿児島大学大学院医歯学総合研究科神経病学講座の髙嶋博先生,医療法人つくし会 南国病院の古谷博和先生,愛媛大学大学院医学系研究科脳神経内科・老年医学講座の三浦史郎先生,済生会松山病院脳神経内科の先生方に深謝申し上げます.
Notes
本報告の要旨は,第113回日本神経学会中国・四国地方会で発表し,会長推薦演題に選ばれた.
文献
- 1) 高嶋博.遺伝性運動性・感覚性・自律神経性ニューロパチーの臨床.臨床神経 2014;54:957-959.
- 2) 清水潤.Hereditary sensory and autonomic neuropathy.Peripheral Nerve 2013;24:23-30.
- 3) Rotthier A, Baets J, Timmerman V, et al. Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat Rev Neurol 2012;8:73-85.
- 4) Moghadam KK, Pizza F, La Morgia C, et al. Narcolepsy is a common phenotype in HSAN IE and ADCA-DN. Brain 2014;137:1643-1655.
- 5) Klein CJ, Botuyan MV, Wu Y, et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet 2011;43:595-600.
- 6) Yuan J, Higuchi Y, Nagado T, et al. Novel mutation in the replication focus targeting sequence domain of DNMT1 causes hereditary sensory and autonomic neuropathy IE. J Peripher Nerv Syst 2013;18:89-93.
- 7) 渡部真志,松本雄志,岡本憲省ら.前頭葉症状で初発したhereditary sensory and autonomic neuropathy 1E(HSAN1E)の1例.臨床神経 2017;57:753-758.
- 8) Klein CJ, Bird T, Ertekin-Taner N, et al. DNMT1 mutation hot spot causes varied phenotypes of HSAN1 with dementia and hearing loss. Neurology 2013;80:824-828.
- 9) Baets J, Duan X, Wu Y, et al. Defects of mutant DNMT1 are linked to a spectrum of neurological disorders. Brain 2015;138:845-861.
- 10) Zheng W, Yan Z, He R, et al. Identification of a novel DNMT1 mutation in a Chinese patient with hereditary sensory and autonomic neuropathy type IE. BMC Neurol 2018;18:174.
- 11) Catania A, Peverelli L, Tabano S, et al. DNMT1-complex disorder caused by a novel mutation associated with an overlapping phenotype of autosomal-dominant cerebellar ataxia, deafness, and narcolepsy (ADCA-DN) and hereditary sensory neuropathy with dementia and hearing loss (HSN1E). Neuro Sci 2019;40:1963-1966.
- 12) Coelho P, Oliveira Santos M. First Portuguese patient presenting with hereditary sensory and autonomic neuropathy type 1E associated with a novel mutation in DNMT1 gene. Neurol Sci 2020;41:1289-1290.
- 13) Parissis D, Christodoulou K, Kleopa KA. Novel de novo DNMT1 gene mutation associated with hereditary sensory and autonomic neuropathy 1E (HSAN1E). Neurol Sci 2023;44:2199-2201.
- 14) Kara E, Tucci A, Manzoni C, et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain 2016;139:1904-1918.
- 15) Hojo K, Imamura T, Takanashi M, et al. Hereditary sensory neuropathy with deafness and dementia: a clinical and neuroimaging study. Eur J Neurol 1999;6:357-361.
- 16) Hojo K, Kawamata T, Tanaka C, et al. Inflammatory glial activation in the brain of a patient with hereditary sensory neuropathy type 1 with deafness and dementia. Neurosci Lett 2004;367:340-343.