Abstract
Amyotrophic lateral sclerosis (ALS) is an adult-onset intractable motor neuron disease characterized by selective degeneration of cortical neurons in the frontotemporal lobe and motor neurons in the brainstem and spinal cord. Impairment of these neural networks causes progressive muscle atrophy and weakness that spreads throughout the body, resulting in life-threatening bulbar palsy and respiratory muscle paralysis. However, no therapeutic strategy has yet been established to halt ALS progression. Although evidence for clinical practice in ALS remains insufficient, novel research findings have steadily accumulated in recent years. To provide updated evidence-based or expert consensus recommendations for the diagnosis and management of ALS, the ALS Clinical Practice Guideline Development Committee, approved by the Japanese Society of Neurology, revised and published the Japanese clinical practice guidelines for the management of ALS in 2023. In this guideline, disease-modifying therapies that have accumulated evidence from randomized controlled trials were defined as “Clinical Questions,” in which the level of evidence was determined by systematic reviews. In contrast, “Questions and Answers” were defined as issues of clinically important but insufficient evidence, according to reports of a small number of cases, observational studies, and expert opinions. Based on a literature search performed in February 2022, recommendations were reached by consensus, determined by an independent panel, reviewed by external reviewers, and submitted for public comments by Japanese Society of Neurology members before publication. In this article, we summarize the revised Japanese guidelines for ALS, highlighting the regional and cultural diversity of care processes and decision-making. The guidelines cover a broad range of essential topics such as etiology, diagnostic criteria, disease monitoring and treatments, management of symptoms, respiration, rehabilitation, nutrition, metabolism, patient instructions, and various types of care support. We believe that this summary will help improve the daily clinical practice for individuals living with ALS and their caregivers.
Introduction
Amyotrophic lateral sclerosis (ALS) is an adult-onset intractable motor neuron disease. Although several medications, including riluzole, edaravone, and sodium phenylbutyrate-taurursodiol, are available globally, their effects are limited, and other treatments only provide symptomatic relief. Several non-pharmacological interventions, such as a high-calorie diet and noninvasive ventilation (NIV), have been reported to be beneficial in slowing the progression of ALS. However, this information is not readily accessible to patients and their families, thereby leaving unmet medical needs. Multidisciplinary approaches are urgently required for practical management of this issue. Clinical practice guidelines for ALS have been issued by global organizations and expert panels1)–6), providing consensus approaches to medication and invasive interventions, including percutaneous endoscopic gastrostomy (PEG) and tracheostomy invasive ventilation (TIV). However, individual decision-making may also depend on cultural and religious diversities, which could be underestimated and potentially explain the seemingly inconsistent efficacy of medications and interventions worldwide.
In May 2023, the Japanese Society of Neurology (JSN) published the latest version of clinical practice guidelines for ALS in Japanese. These guidelines cover a wide range of topics, including etiology, diagnostic criteria, disease progression monitoring, symptom management, nutritional support, respiratory care, and end-of-life care. This article summarizes in English the Japanese guidelines available on the JSN website (www.neurology-jp.org/guidelinem/). This guideline provides knowledge to undstand ALS to improve the quality of life of patients and their families. The guideline is a valuable resource for healthcare providers and caregivers to improve the quality of life for patients and their families in Japan. We believe that disseminating the contents of this guideline to the rest of the world will provide useful options for improving the quality of life of ALS patients after diagnosis. In this English version, we aimed to provide the minimal essence of the regional guidelines to offer evidence-based recommendations for the diagnosis, treatment, and management of ALS, highlighting regional and cultural diversities of care processes and decision-making, especially regarding invasive interventions such as PEG and TIV.
The guideline panel comprised multidisciplinary experts, including neurologists, nurses, rehabilitation therapists, medical social workers, caregivers, and patients from the Japan ALS Association. An independent team conducted a systematic review (SR) of the literature to provide evidence for recommendations. We believe that this article may contribute to a better understanding of the regional and cultural diversity of ALS and provide evidence-based recommendations for clinical practice.
Outlines of ALS
Epidemiology and subtypes
The incidence of ALS in Japan is estimated to be 2.2/100,000 persons/year, with a prevalence of 9.9/100,000 persons/year7). The incidence rate increases with age, peaking in the 70s and decreasing in the 80s. Males are 1.3 to 1.5 times more likely to develop the disease than females.
ALS can be classified into different types based on several factors. Most ALS cases are sporadic, and approximately 5–10% of ALS cases are hereditary, known as familial ALS (FALS). Known causative genes are found in approximately 50% of the FALS cases. In Japan, SOD1 mutations account for 32–36% of FALS cases, followed by FUS in 8–11% and TARDBP in 2–3% of cases. C9ORF72 is the most common causative gene in Europe and the United States but is extremely rare in Asians, including the Japanese. Even among patients with sporadic ALS, there are cases of causative genes for FALS8)9).
Classification according to the site of initial onset is divided into the spinal (classical) and bulbar types. The spinal type is usually characterized by muscle atrophy and weakness in one limb, with a course that spreads to adjacent sites and other areas. The classical type is narrowly defined as the beginning of muscle atrophy distal to the upper limbs. The bulbar type begins with dysarthria and/or dysphagia due to bulbar palsy and spreads to the neck muscles, shoulder girdle muscles, and muscles of the four limbs. In the early stages, symptoms may be limited to bulbar palsy, and the motor skills of the limbs may be preserved for some time. Such cases are referred to as progressive bulbar palsy. Rarely, the disease begins with paralysis of the respiratory muscles and is called the respiratory muscle type.
ALS is characterized by progressive degeneration of both upper and lower motor neurons but may be clinically dominated by either upper or lower motor neuron syndromes. When there are clinically progressive upper motor neuron syndromes but no lower motor neuron syndromes, the disease is called the upper motor neuron or pyramidal type. When these criteria are met, such as the absence of lower motor neuron syndromes and the presence of progressive upper motor neuron syndromes in two or more upper limbs, lower limbs, or bulbar regions, a diagnosis of primary lateral sclerosis (PLS) can be made > 2 years after disease onset10). While the opinion exists that PLS is an independent disease, there is a strong opinion that PLS is a form of ALS, because lower motor neuron syndromes can appear later, and autopsy pathology may show degeneration of lower motor neurons, even if PLS is clinically diagnosed. When clinically progressive lower motor neuron syndrome is present but upper motor neuron syndrome is absent, it is referred to as progressive muscular atrophy (PMA). PMA, which is limited to both upper limbs, is called the flail arm type, while brachial amyotrophic diplegia, man-in-the-barrel type, and Vulpian-Bernhardt subtype are also of the same type. Autopsy pathology of PMA often shows degeneration of the upper motor neurons, most of which are thought to be ALS.
Clinical course and prognosis
ALS symptoms are progressive, and in many cases, muscle weakness spreads from the initial site to adjacent sites. As the disease progresses, patients have difficulty with independent activities of daily living, such as personal hygiene, grooming, transfers, feeding, and elimination. Dysphagia due to bulbar palsy increases the risk of malnutrition and aspiration pneumonia. Respiratory muscle paralysis can lead to dyspnea and respiratory failure. During the course of the disease, various non-motor symptoms such as cognitive decline (mainly frontotemporal lobe dysfunction), pseudobulbar affect, pain in various parts of the body, depression, fatigue, sleep disturbance, painful muscle cramps, drooling, and constipation may occur.
The median time from disease onset to death or induction of invasive ventilatory support has been reported to be 32–48 months11)12) in Japanese patients with ALS. However, the progression of symptoms and prognosis in individual patients vary considerably. Older age at onset, the onset of bulbar palsy, the onset of respiratory impairment, concomitant frontotemporal dementia, a greater rate of decline on the functional rating scale, early spread of symptoms to multiple body regions, and early and significant weight loss are associated with shorter survival from onset13)–15).
TIV has been shown to significantly prolong the survival of patients with ALS. A report from a Japanese multicenter ALS patient registry16) showed that TIV prolonged the median survival of patients by 6.7 years in a TIV-treated group compared with a background-matched non-TIV group, with the survival benefit being greater in younger patients than in older patients. Communication difficulties17) and complications such as dysuria, macroglossia, hypothermia, unstable blood pressure, hyperglycemia, and various infections are crucial issues that must be addressed in life after ventilator support18).
Recommendations from expert consensus
Diagnosis of ALS
ALS is diagnosed based on (1) the presence of upper and lower motor neuron dysfunction, (2) progressive disease course, and (3) differential diagnosis. Since there is no biochemical biomarker, diagnosis is made by combining clinical findings and supplemental tests, such as electrophysiology and imaging (Table 1, Boxes 1 and 2).
Table 1 Diagnosis of amyotrophic lateral sclerosis.
| Subject |
Recommendations |
| Diagnostic criteria of ALS |
Several diagnostic criteria have been proposed to increase diagnostic sensitivity. The El Escorial criteria were initially proposed and revised to improve diagnostic sensitivity. Its modification in electrodiagnostic criteria was proposed in the Awaji criteria and updated Awaji criteria40). Complexity of the diagnostic scheme of these criteria may result in low reproducibility. In 2019, the Gold Coast criteria were proposed to simplify diagnostic processes, and their validation has been expected41). |
| Differential diagnosis |
Other diseases must be excluded for diagnosis of ALS. A suggested list of differential diagnosis is shown in Box 1 and 2. |
| What to expect when only fasciculations are present |
Long-term observation is needed because progression to ALS in the future cannot be excluded. |
| Significance of electrophysiology |
Needle electromyography is important for the detection of lower neuron abnormality. Nerve conduction study is necessary to exclude demyelinating neuropathy. |
| Selection of muscles to perform electromyography |
First, divide the body into four segments (cranial, cervical, thoracic, and lumbosacral). At least one muscle in the cranial and thoracic segments is selected, whereas at least two muscles are selected in the cervical and lumbosacral segments that are differently innervated by the levels of the peripheral nerves and nerve roots. |
| Evaluation of respiratory muscles |
In addition to disease history, symptoms, and physical examination, objective tests, such as forced vital capacity, saturation of percutaneous oxygen, peak cough flow, end-tidal carbon dioxide, overnight measurement of SpO2, and arterial blood gas measurement, are utilized. |
| Utilization of imaging study for diagnosis |
To date, no imaging studies have enabled the diagnosis of ALS. MRI of the brain and spinal cord is required to exclude other diseases. |
| Evaluation of upper motor neuron dysfunction |
No test has been established as diagnostically necessary. Several tests have been studied, such as prolongation of central motor conduction time measure by transcranial magnetic stimulation, decreased amplitudes of motor evoked potentials, impairment of short-interval intracortical inhibition, and abnormal signal intensity of the motor cortex and pyramidal tract on MRI. |
| Role of blood and cerebrospinal fluid tests in diagnosis |
Blood and cerebrospinal fluid tests are important to rule out other diseases in the diagnosis of ALS, but specific blood and cerebrospinal fluid markers indicative of ALS have not yet been established. |
| Significance of the split-hand phenomenon |
The split-hand phenomenon refers to the different degrees of muscle atrophy between the impaired thenar and the first dorsal interosseus muscles and the relatively preserved hypothenar muscles. This finding is highly specific to ALS. |
| Significance of neuromuscular ultrasound |
Peripheral nerve ultrasound may detect the thinness of peripheral nerves and nerve roots, although its diagnostic sensitivity is low in early ALS. Muscle ultrasound may detect denervated changes and fasciculations. Current diagnostic criteria do not require sonographic findings; thus, their diagnostic significance needs to be established. |
ALS, amyotrophic lateral sclerosis; MRI, magnetic resonance imaging.
Box 1 Diagnostic process for amyotrophic lateral sclerosis.
| Requirement of diagnosis of amyotrophic lateral sclerosis |
| A. Presence of the following: |
| 1. Clinical or electrophysiological evidence of lower motor neuron dysfunction |
| 2. Clinical evidence of upper motor neuron dysfunction |
| 3. Progressive disease course and spread to other body parts |
| B. Exclusion of other diseases: |
| 1. Electrophysiological or pathological evidence of other diseases to explain clinical findings |
| 2. Imaging abnormality to explain clinical or electrophysiological findings |
Box 2 Differential diagnosis of amyotrophic lateral sclerosis.
| • Motor neuron diseases |
| Spinal muscular atrophy |
| Spinal and bulbar muscular atrophy |
| Post-polio syndrome |
| Hexosaminidase A deficiency |
| Hereditary spastic paraparesis |
| • Peripheral neuropathies/radiculopathies |
| Cervical and lumbosacral radiculopathies |
| Multifocal motor neuropathy |
| Chronic inflammatory demyelinating polyneuropathy |
| Cramp-fasciculation syndrome |
| Neuromyotonia |
| Charcot–Marie–Tooth disease |
| Paraneoplastic syndrome |
| Heavy metal poisoning |
| Mononeuritis multiplex |
| Neurofibromatosis |
| Hereditary motor and sensory neuropathy with proximal dominant involvement |
| • Neuromuscular junction diseases |
| Myasthenia gravis |
| Lambert-Eaton myasthenic syndrome |
| • Brain and spinal cord diseases |
| Syringomyelia |
| Multiple sclerosis |
| Hirayama disease (monomelic amyotrophy) |
| Lyme disease |
| HIV/HTLV-1 infection |
| Stroke |
| Basal ganglia disorders |
| • Myopathies |
| Inclusion body myositis |
| Inflammatory myopathy |
| Adult polyglucosan body disease |
| Facial onset sensory and motor neuronopathy syndrome |
| • Endocrine diseases/ nutritional conditions |
| Hyperthyroidism/ Hyperparathyroidism |
| Subacute combined degeneration of spinal cord |
| Celiac disease |
Diseases of significant importance are underlined. HIV, human immunodeficiency virus; HTLV-1, human T-cell leukemia virus type 1.
Two anti-ALS drugs, riluzole and edaravone19), are available in the United States and Japan (as of December 2022). Strong opioids and several muscle relaxants have been approved for use by the Japanese government to relieve respiratory distress, pain, and spasticity in patients with ALS. Based on the unavailability of drugs in Japan, the current version of the practice guidelines focuses on riluzole, edaravone, and strong opioids, for which standard protocols of their use should be indicated. To determine the recommendation levels according to the Grading of Recommendations Assessment, Development and Evaluation (GRADE) system, only riluzole and edaravone were selected because they have relatively high levels of evidence, indicating P: population, I: intervention, C: comparison, and O: outcomes (PICO) conducted by randomized controlled trials (RCTs). In this guideline, patients with ALS are designated as ‘P,’ while ‘I’ refers to drugs or care, and ‘C’ denotes a placebo. Additionally, the guideline selected five outcomes: (1) inhibition of ALS Functional Scale-Revised (ALSFRS-R) decline, (2) extension of non-tracheostomy survival, (3) reduction in respiratory function decline, (4) occurrence of serious adverse events, and (5) improvement in quality of life (QOL). After a thorough review, only riluzole and edaravone were deemed to have evaluable evidence (Table 2).
Table 2 Medications.
| Treatments |
Recommendations |
| Approved medications |
• Riluzole and edaravone are covered by insurance for ALS treatment. |
| • Strong opioids are approved by the insurance coverage for managing pain and respiratory distress in patients with ALS. |
| • Muscle relaxants are approved by insurance coverage for managing spasticity in patients with ALS. |
| Riluzole |
• Riluzole is recommended for patients with ALS (GRADE 2B). |
| • There is strong evidence that riluzole extends survival until tracheostomy invasive ventilation. However, evidence is limited regarding the beneficial effect in delaying the functional decline. |
| • Common side effects of riluzole include dizziness, nausea, and anxiety. Severe liver dysfunction is a contraindication for using riluzole. |
| Edaravone |
• Edaravone is recommended for patients with ALS (GRADE 2B). |
| • Evidence is limited regarding the beneficial effect in delaying the functional decline, and no evidence for extending the survival is provided. |
| • Severe renal dysfunction is a contraindication. Serum cystatin C level should be monitored to assess renal function as creatine kinase may be underestimated owing to muscle atrophy. |
| Opioid |
• Morphine hydrochloride hydrate is initially administered based on patients’ complaints, and subsequently, it is gradually switched to morphine sulfate hydrate (Fig. 1). |
| • In the end-of-life stages, continuous intravenous or subcutaneous infusion of morphine hydrochloride may be utilized. |
| • The adverse effects of opioids should be carefully monitored. |
| Alternative therapies |
• If the patient/family asks about alternative therapies, the fact that there is no medical evidence that alternative therapies are effective for ALS should be explained. |
| • As patients often use alternative therapies voluntarily and these therapies may pose a direct or indirect risk to their health, the healthcare provider should confirm the use of alternative therapies with the patient. |
| Disease-modifying therapies |
• Currently, only riluzole and edaravone have been approved, and many agents are in trial phases for their disease-modifying effects. |
| • Such drugs are not recommended for use outside of clinical trials because their efficacy and safety are not validated. |
| Regenerative therapies |
The current evidence is insufficient to recommend regenerative therapies for patients with ALS. |
ALS, amyotrophic lateral sclerosis; GRADE, The Grading of Recommendations Assessment, Development and Evaluation.
Riluzole: Riluzole is a widely recommended medication for the treatment of ALS. It attenuates excitotoxicity caused by the presynaptic release of glutamate and prolongs the survival of patients or the time to TIV dependence by 3 months. Based on an SR of high-grade evidence, the guideline committee recommends the use of riluzole under certain conditions (GRADE 2B). However, patients and their families must be informed that although the evidence levels are high, riluzole has limited effectiveness in extending survival without TIV. Additionally, evidence regarding the inhibition of motor decline, as estimated using the ALSFRS-R, is scarce. Patients must be closely monitored for serious side effects, such as liver dysfunction and interstitial pneumonia.
Edaravone: Edaravone is an antioxidant that is recommended for the treatment of ALS. Four RCTs demonstrated PICO. The guideline committee recommends the use of edaravone under certain conditions (GRADE 2B). Although edaravone has been shown to slow the functional decline estimated by the ALSFRS-R, evidence regarding its efficacy in extending survival is lacking. Moreover, renal and liver dysfunction should be monitored as an adverse effect.
Symptomatic management
In addition to amyotrophy and muscle weakness, patients with ALS may experience spasticity, painful muscle cramps, sialorrhea associated with bulbar palsy, and breathlessness owing to respiratory muscle involvement. Moreover, a variety of non-motor symptoms, such as cognitive decline, mood disorders, pseudobulbar affect, sleep disturbances, fatigue, and pain, may occur as initial symptoms. However, each symptom may not occur in all patients. Constipation is a commonly reported comorbidity, and pressure ulcers can develop in patients with ALS. In daily clinical practice, it is necessary to inquire about symptoms that are negatively related to the QOL, determine the cause, and provide optimal care and symptomatic treatment. Non-pharmacological therapies are primarily considered to avoid non-essential medications or polypharmacy (Table 3). Although evidence for symptomatic treatment is currently scarce, elucidation of the underlying pathology and intervention research is required, particularly for non-motor symptoms of ALS.
Table 3 Symptomatic Management.
| Symptoms |
Recommendations |
| Cognitive impairment42) |
• To distinguish frontotemporal cognitive impairment associated with ALS from other coexisting dementia, such as Alzheimer’s disease, repeated assessments from the time of ALS diagnosis are recommended. |
| • Sudden onset, exacerbation, or fluctuation in cognitive impairment may indicate delirium, and physical complications (including respiratory failure) should be ruled out. |
| Anxiety and depression43) |
• First, we recommend mental health care and support through multidisciplinary medical care. |
| • Second, if medication is required for anxiety, consider SSRIs, SNRIs, or anxiolytics with attention to respiratory inhibition. Low-dose trials of SSRIs, SNRIs, and tricyclic antidepressants may be considered for depression. |
| Pseudobulbar affect44) |
• Ask patients whether they have uncontrollable, sudden crying or laughing symptoms that are unrelated to emotion. |
| • Encourage patients and their caregivers to understand the symptoms and assess the degree of distress. |
| • If the symptoms are painful for the patient, consider administering an SSRI or an SNRI at a low dose (both of which are not covered by insurance in Japan). |
| Insomnia45) |
• Clarify the cause and pathology of insomnia and ensure the quality of sleep. |
| • Beware of insomnia due to hypoventilation during sleep. |
| • NIV is effective for hypoventilation during sleep but only for a certain period. |
| Fatigue46) |
• Fatigue that does not fully recover, even with rest, is common in patients with ALS, but symptomatic treatment has not been established. |
| • Evaluate respiratory function because of possible association between fatigue and early respiratory distress47). |
| Pain46)48)49) |
• Identify the cause and pathology of pain and actively alleviate it. |
| • Initiate nonsteroidal analgesics as needed for musculoskeletal nociceptive pain, including painful muscle spasms, spasticity, contractures, immobility, and pressure. |
| • Consider early introduction of opioids in refractory cases and patients with poor QOL. |
| Breathlessness46)49) |
• Identify the cause of breathlessness and actively alleviate it. |
| • First, differentiate common respiratory illnesses from respiratory symptoms associated with ALS that may be mitigated by NIV but for a certain period. |
| • For dyspnea in a patient on a ventilator, after checking the entire circuit, improve airway clearance and optimize settings using monitoring data. |
| • If NIV is undesirable or if NIV is already introduced but difficult to optimize, consider early opioid introduction and/or low-dose oxygen. |
| Spasticity49) |
• Prevent joint contractures using physical therapy, such as passive exercise, and consider antispasmodics as needed. |
| • Consider intrathecal baclofen therapy with caution for severe and persistent spasticity that cannot be adequately controlled with oral medications. |
| Sialorrhea46) |
• Anticholinergic drugs, such as tricyclic antidepressants, may require caution owing to their adverse effects. |
| • Low-pressure continuous saliva aspiration devices may also be useful but are not approved as medical devices in Japan. |
| Constipation46) |
• Note that patients with ALS are prone to constipation, and actively check it. |
| • Manage the concomitant constipation according to general prophylaxis and treatment. |
| Pressure ulcers |
• Recognize the risk of developing pressure ulcers in areas of pressure caused by immobility, and actively prevent pressure ulcers by focusing on decompression, skin care, and nutritional management. |
| • Treat pressure ulcers according to general recommendations. |
ALS, amyotrophic lateral sclerosis; NIV, noninvasive ventilation; QOL, quality of life; SNRI, serotonin norepinephrine reuptake inhibitor; SSRI, selective serotonin reuptake inhibitor.
Opioids can be used to relieve symptoms, such as dyspnea, pain, restlessness, and burning sensation. They should be initiated in the early stages of suffering whenever possible. In Japan, only morphine sulfate hydrate and morphine hydrochloride hydrate are permitted for use in patients with ALS. Morphine, when appropriately used in ALS clinics, does not exacerbate hypercapnia and hypoxia or accelerate death20). Initially, morphine hydrochloride hydrate (a short-acting drug) should be administered at a dose of 2.5 mg at a time (Fig. 1)2). If this dose is well-tolerated, it may be increased to 5 or 10 mg per administration. Subsequently, based on patient complaints, multiple doses can be administered every 3–4 h. When the total daily dose of morphine hydrochloride exceeds 10–20 mg, it may be switched to morphine sulfate hydrate (a long-acting drug), which is administered twice daily. Rescue doses of morphine hydrochloride can also be added as needed, based on patient complaints. In the end-of-life stages, continuous intravenous or subcutaneous infusion of morphine hydrochloride may be initiated to stabilize the effect of relieving suffering21).

Respiratory impairment is the most important life-threatening symptom of ALS. In recent years, respiratory management has evolved remarkably, with the early identification of respiratory dysfunction, systematization of airway clearance techniques, and widespread use of a variety of assisted ventilation therapies. However, evidence and recommendations for effective respiratory therapy in ALS are lacking, owing to the lack of large RCTs. Here, we summarized the current status of respiratory management in ALS based on articles and literature used to develop guidelines. Additionally, we noted that Japan is unique in the implementation of long-term ventilatory therapy and discussed the effectiveness and complications of this therapy (Fig. 2, Table 4).
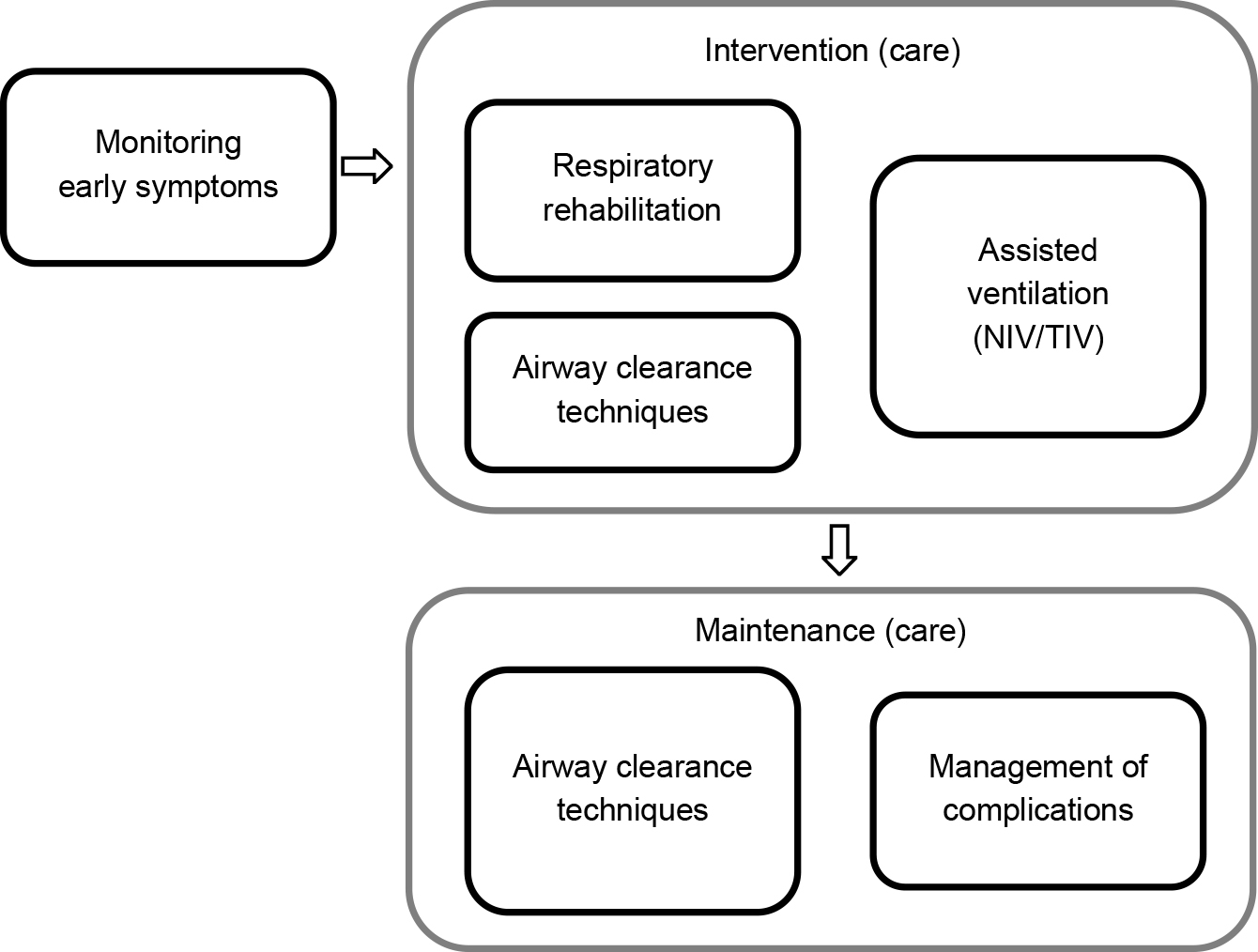
Table 4 Respiratory management.
| Subject |
Recommendations |
| Early identification of respiratory dysfunction |
• Because respiratory dysfunction occurs before the onset of respiratory symptoms, periodic tests of FVC should be performed50). |
| • Nocturnal pulse oximetry and polysomnography are used to detect nocturnal hypoventilation before FVC decreases. |
| Respiratory rehabilitation for ALS |
Rehabilitation for respiratory dysfunction may affect patient survival; therefore, inspiratory muscle training, lung volume recruitment training, and MAC are commonly used. It should be initiated before the onset of respiratory symptoms. |
| Airway clearance managements (expectorant methods) |
• Central airway clearance (cough augmentation) methods include MAC, inhalation assistance using a bag-bubble mask, and MI-E51). |
| • Peripheral airway clearance (moving secretions to the central side) methods include manual techniques, high-frequency chest wall compression, MI-E with high-frequency oscillations, and intrapulmonary percussive ventilation51). |
| Ventilation |
NIV and TIV are available. The effectiveness of diaphragmatic pacing has been denied. |
| Benefits and problems of NIV |
• There is insufficient evidence to support the efficacy of NIV. Observational studies suggest that NIV not only improves respiratory symptoms but also (1) prolongs non-tracheostomy survival, (2) improves and maintains quality of life, (3) improves sleep quality, and (4) reduces decline in respiratory function52). |
| • Problems include discomfort associated with the use of the device, skin problems, and aerophagy. In the advanced stages, upper airway obstruction due to aspiration of saliva and tongue deposition becomes difficult to manage, and respiratory symptoms may not be relieved by NIV alone. |
| NIV — When to start and set up53) |
• Initiate at the latest when clinical signs of respiratory compromise are observed. |
| • Initiating NIV after the onset of respiratory symptoms may relieve symptoms. Early initiation of NIV may prolong survival53). |
| • No specific ventilation mode has been shown to be superior in the NIV setting. However, several observational studies have suggested the use of VTPCV with a target volume of approximately 8 ml/kg body weight54). |
| • After initiation of NIV, periodic adjustments are necessary as the patient’s symptoms progress. |
| • Patients/caregivers must be actively involved in the initiation, maintenance, and management of NIV therapy and must be held accountable for their decisions. |
| • Evaluate the effectiveness of NIV therapy, and modify the setting as respiratory dysfunction progresses. Bulbar dysfunction, drooling, difficulty coughing up airway secretions, cognitive impairment, and inappropriate interface are barriers to continued NIV therapy and should be appropriately evaluated and addressed. |
| TIV — When and how to initiate and complications in patients with long-term TIV |
• Although there are no universally accepted guidelines regarding the criteria for initiation of TIV, it should be performed when the decision to induction has been made, SpO2 > 90% and pCO2 < 50 mmHg cannot be maintained even with appropriate use of NIV and MI-E, and secretions cannot be adequately managed55). |
| • Because of the many problems that occur when TIV therapy is initiated on an emergency basis, it is preferable to schedule the introduction of TIV therapy on a standby basis. |
| • Although there is no recommended TIV setting for ALS, volume-controlled settings such as volume-controlled ventilation or VTPCV are commonly used. The tidal volume should be set at approximately 8 ml/kg standard body weight56). |
| • Non-motor symptoms include an enlarged tongue (macroglossia), autonomic dysfunction, glucose intolerance, hypothermia, and dysuria. Complications of long-term TIV therapy include infection, cholelithiasis, urinary tract stones, and bedsores18). |
ALS, amyotrophic lateral sclerosis; FVC, forced vital capacity; MAC, manually assisted cough; MI-E, mechanical insufflation-exsufflation, NIV, noninvasive ventilation; TIV, tracheostomy invasive ventilation; VTPCV, volume-targeted pressure-controlled ventilation.
The major goal of rehabilitation medicine is the recovery from functional disability. However, in diseases such as ALS, which are characterized by progressive muscular weakness throughout the body and for which curative treatment is unavailable, the rehabilitation goal is to maintain the remaining abilities or allow maximum performance of activities using assistive devices, ultimately improving the QOL of patients and their families22)23) (Table 5, Box 3).
Table 5 Rehabilitation medicine in amyotrophic lateral sclerosis.
| Subject |
Recommendations |
| Physical therapy and exercise57) |
• Range of motion exercises, body position change training, and activities of daily living practice are performed. |
| • Moderate resistance training and aerobic exercise may be temporarily effective for muscles with mild to moderate weakness. |
| • Excessive exercise load may worsen muscle weakness; therefore, be careful not to cause muscle pain, fatigue, respiratory symptoms, or worsening the next day. |
| • The wearable cyborg robot, Hybrid Assistive Limb®, has the potential to extend the walking distance and reconstruct the walking system in cases of ALS58). |
| Dysarthria59)60) |
• Although effective rehabilitation techniques to prevent the progression of speech disorders have not been established, support for communication disorders can be provided through instructing augmentative and alternative communication. |
| Assistive devices (aids equipment and adaptations)61) |
• Assistive devices can be useful in providing assistance with daily activities, mobility, and communication with others (Box 3). |
| Quality of life |
• Physical therapists, occupational therapists, and speech-language pathologists provide rehabilitation treatment according to the needs of the patient, and the intervention of clinical psychologists can also contribute to maintaining the patient’s QOL62). |
| • Noninvasive ventilation therapy has the potential to improve the QOL of patients63). |
| • Multidisciplinary team care provided from the early stage of disease can improve the QOL of patients, and patient care and caregiver approaches can improve the QOL of caregivers64). |
| • Caregiver burden can affect the QOL of caregivers65); therefore, it is important to provide appropriate care and services that caregivers need. |
| • There are multiple QOL assessment scales for patients with ALS. The choice depends on whether the purpose is to evaluate the effectiveness of therapeutic or care interventions or to evaluate the QOL in terms of clinical symptoms, health status, and satisfaction with life. |
| Sexual behavior66) |
• If there is an agreement between the patient and their spouse/partner, there is no need to impose restrictions on sexual activity. Pregnancy and childbirth are also possible, but it is necessary to consider the potential impact on the mother’s body and fetus as well as the possibility of ventilator use. |
ALS, amyotrophic lateral sclerosis; QOL, quality of life.
Box 3 Assistive devices, welfare equipment, and home improvement for amyotrophic lateral sclerosis.
| Assistive devices |
Welfare equipment |
Home improvement |
| Orthotics, brace |
Aid for eating and drinking |
Hand-rails |
| Wheelchair |
Bed and detachable bed board with powered adjustment |
Lifting platform |
| Electric wheelchair |
Pressure ulcer prevention device |
Ramped entrance |
| Walker |
Apparatus for position change |
Slip resistance |
| Walking aid (e.g., stick) |
Transfer lift |
Change to sliding door |
| Computer-based communication system |
Bathroom aids |
Improvement of lavatory |
|
Communication aid (calling device, operating switch, handwriting communication aid, communication board) |
|
ALS exhibits characteristic metabolic and nutritional dysfunction in both the early and advanced stages with invasive ventilation. Progressive weight loss, anorexia, and hypermetabolism are specific phenomena associated with ALS24)25). Weight loss and hypermetabolism have been established as prognostic factors for short survival. Recent discussions on metabolic dysfunction in ALS have focused on hypothalamic lesions and the shift in fuel utilization toward lipid metabolism. The effect of a high-calorie diet (rich in fats) on survival has been reported26), and preventing weight loss after diagnosis is expected to improve the survival prognosis. Nutritional intervention holds promise as a disease-modifying therapy for ALS, and formulae have been reported for calculating the ideal energy intake in patients with ALS27)28). Gastrostomy tube insertion is an effective approach for implementing nutritional interventions, and the optimal timing of insertion should be determined based on clinical monitoring of the patient’s body weight, swallowing function, and respiratory function29)30). However, nutritional supplements or drugs that effectively control body weight have not been identified. Following the initiation of invasive ventilation, the patient’s metabolic rate progressively decreases31), and adverse manifestations of hypometabolism may arise at an advanced stage18). Energy intake should be restricted based on the nutritional status of the patient at this late stage (Table 6).
Table 6 Nutritional and metabolic dysfunction in amyotrophic lateral sclerosis.
| Subject |
Recommendations |
| Clinical significance of nutritional and metabolic disturbance |
Weight loss and reduction rate of weight in the early stage are independent prognostic factors for short survival24). In addition to muscle wasting and decreased energy intake, disease-specific hypermetabolism may play a crucial role in weight loss25). Weight loss in the early stage also predicts functional prognosis during the advanced stage with invasive ventilation. In the stages with invasive ventilation, hypometabolism and weight gain may occur31). Hypothalamic lesions have been reported as candidates for metabolic dysfunctions. |
| A protective role of low-density lipoprotein cholesterol and an adverse prognostic effect of high-density lipoprotein cholesterol have been reported, but they are yet to be established67). |
| Nutritional management in the early stage |
A high calorie diet, particularly containing high fat, is recommended to maintain body weight and prolong survival period26). Two formulae have been reported for calculating the ideal energy intake for Japanese patients in the early stage, which are as follows27)28): |
(1) TEE = 1.68 × BEE + 11.8 × ALSFRS-R − 690
where, TEE: total energy expenditure (kcal/day)
BEE: basal energy expenditure calculated by Harris-Benedict formula (kcal/day)
ALSFRS-R: total score of the Revised ALS Functional Rating Scale. |
(2) REE = 1.000251 × BEE + 313.3507 × TV − 112.036
where, REE: resting energy expenditure (kcal/day)
BEE: basal energy expenditure calculated by Harris-Benedict formula (kcal/day)
TV: tidal volume (L)
TEE = REE × activity factor |
| Assessment of swallowing function |
Swallowing function should be regularly monitored through patients’ symptoms, physical examinations, dysphagia screening tests, videofluoroscopy, and videoendoscopy. Dysphagia screening tests include repetitive saliva swallowing test, modified water swallowing test, and cervical auscultation. Measurements of tongue pressure and thickness are also valuable. Additionally, assessments of the bulbar function, nutritional state, and respiratory function are necessary. |
| Management of dysphagia |
Direct and indirect swallowing rehabilitation by a multidisciplinary team is required. Direct swallowing rehabilitation involves training with real food. Indirect swallowing rehabilitation includes exercises such as jaw opening exercises, head raising exercises, blowing exercises, Mendelsohn maneuver, and others. Expiratory strength training is also recommended to maintain the swallowing function. When the risk of choking and aspiration arises, feeding via a gastric tube should be considered, and aspiration prevention surgery may be an alternative approach. |
| Intervention for patients who cannot eat perorally |
In patients at risk for aspiration and experiencing progressive weight loss, feeding via a nasogastric or gastrostomy tube should be considered to prevent aspiration. Parenteral nutrition may be considered for patients who have gastrointestinal problems or prefer not to be fed through nasogastric or gastrostomy tubes. |
| Indication of PEG |
The indications for PEG are as follows: (1) >10% reduction in weight from premorbid weight29), (2) early symptoms of dysphagia, including decreased food intake, prolonged ingestion time, and choking, and (3) preserved respiratory function (forced vital capacity ≥ 50%)30). It is recommended to perform PEG before arterial carbon dioxide pressure increases. Patients with low vital capacity or those with noninvasive ventilation may undergo PEG with respiratory support using a noninvasive ventilator. Radiologically inserted gastrostomy is not popular in Japan. The main risk associated with PEG is the deterioration of respiratory function during or after the procedure. |
| Nutritional management in the advanced stage with invasive ventilation |
In the advanced stage with TIV, hypometabolism typically develops, and energy intake should be restricted28). Specifically, in patients in a totally locked-in state, the daily energy requirement is below 800 kcal/day. Weight gain, subcutaneous and visceral fat accumulation, macroglossia, and a hyperglycemic state may develop in the advanced stage with TIV18). An appropriate nutritional intervention for patients with noninvasive ventilation has not yet been established. |
| Indication, methods, and complications of aspiration prevention surgery |
Aspiration prevention surgery is considered in patients with repetitive aspiration. The surgery results in irreversible loss of vocalization and nasal airflow. Although some patients may experience temporary ability to ingest and taste food after the surgery, restoration of swallowing function is not guaranteed. The operative procedures include total laryngectomy, laryngeal closure, tracheoesophageal diversion, and laryngo-tracheal separation. Close cooperation between a neurologist and otolaryngologist is necessary for decision-making and patient support. The main complications of surgery are wound infection and deterioration of respiratory function. Chronic adverse events include asphyxia due to temporary sealing of a tracheal fistula and aerophagia. Advanced care planning should be discussed before surgery because patients may become permanently dependent on mechanical ventilation after the procedure. |
PEG, percutaneous endoscopic gastrostomy; TIV, tracheostomy and invasive ventilation.
Multidisciplinary care is a system in which physicians (e.g., neurologists, respiratory physicians, gastrostomy physicians, rehabilitation physicians, psychiatrists, and palliative care physicians), specialized nurses, pharmacists, rehabilitation therapists, medical social workers, dieticians, and clinical research coordinators work together to solve problems. Patients with ALS face various difficulties and multidisciplinary care is considered the most effective method1).
Communicating the diagnosis
In general, explain the medical condition to the patient first. Encourage the presence of family members, caregivers, and professionals with the patient’s consent. Avoid explaining the condition to the family or caregivers before explaining to the patient. Explain the entire picture of the disease; take adequate time, and consider the patient’s feelings during explanation32)–34). Specialists from various professionals must provide specific medical and social care services. If necessary, repeat the explanation until understanding and acceptance are achieved. Consider step-by-step explanations based on patient comprehension and acceptance (Box 4).
Box 4 Checklist of things to keep in mind when explaining the patient’s medical condition
68).
| □ Before explaining the patient’s medical condition, prepare the environment and have all patient information at hand, including family structure, medical history, mental and cognitive function, physical examination findings, and test results. |
| □ Allow sufficient time (45–60 min if possible) for explanation and leave the hospital phone with other staff or keep it on silent mode to avoid interruptions. Inform the patient in advance if time is limited. |
| □ With the patient’s consent, it is desirable to have family members, primary caregivers, nurses, medical social workers, care managers, and other professionals in attendance. |
| □ Obtain an understanding of the patient’s current situation and the degree to which he or she wants to know about the disease. |
| □ It is not necessary to provide all the information at once. Explain in detail in several parts according to the pace of the patient’s understanding. |
| □ Be warm, attentive, respectful, honest, and compassionate. Be careful not to be overly sentimental. |
| □ Provide important information first but include information that is difficult for the patient, along with good information. Do not end up providing bad information only because the patient is upset. Adjust the content, amount, and manner of communication based on the patient’s and family’s reactions. |
| □ Seek to understand the patient’s understanding and feelings (e.g., pessimism, unrealistic expectations). |
| □ Use expressions and vocabulary that are easily understood by the patient. |
| □ Do not be overly direct or use expressions that deny possible treatments (e.g., “There is no treatment,” “There is nothing we can do”). |
| □ Allow sufficient time for frequent and sufficient questioning. |
| □ Explain that the overall condition and prognosis of the disease vary from person to person and that the information available on the Internet or in books may not always be true. |
| □ Explain that even if the condition is not curable, there are various ways to improve it. Specifically, explain that treatment is available to alleviate symptoms, care and assistance are available to make life a little easier, complications can be treated, and the patient will be involved responsibly to the end through cooperation with hospitals and other facilities. |
| □ Ensure that everything is done to maintain the patient’s function, but the patient’s decisions regarding treatment are respected. |
| □ If the patient is unable to decide on a treatment plan, do not urge the patient to do so, but identify and explain the obstacles that prevent a decision. Tell the patient that he or she may decide later. |
| □ Tell the patient that even if a treatment plan has been decided upon, it may be changed later. |
| □ Agree that the patient may seek a second opinion if he/she is willing to do so. |
| □ Explain why this method of communication was used. |
| □ Ensure that the multidisciplinary team cares for the upset patient after explaining the medical condition. |
| □ If the patient does not have sufficient understanding or acceptance, repeat the explanation. |
| □ Depending on the patient’s state of understanding and acceptance, consider providing step-by-step explanations. |
Life prognosis should be communicated as early as possible after diagnosis and over time1)35). It should be emphasized that weight maintenance and NIV improve the prognosis. As prognosis varies considerably with or without TIV, it is important to simulate and communicate the means of communication, care, and financial issues that may arise if TIV is introduced. Surgery requiring general anesthesia should be avoided in patients with ALS. Spondylosis surgery in patients with ALS is unlikely to cause improvement and has not been reported to prolong survival36). Even when the respiratory function is not compromised, careful attention should be paid to the respiratory function, degree of progression, prognosis, and whether TIV is desired. Particularly in patients with impaired respiratory function, gastrostomy for nutritional therapy is recommended to be performed under NIV, which reduces safety concern37).
Shared decision-making to collaborative decision-making
Shared decision-making (SDM), which emphasizes the process of decision making, is promoted by sharing information among patients, family members, caregivers, and specialists in various professions. When a patient is unable to decide on his/her own, the team should share ethical viewpoints while referring to the opinions of surrogates who know the patient well35).
End-of-life care issues
“The terminal phase” is considered to be “the time when the patient is prepared for death in the near future,” and the plan of care is discussed while assuming that the patient’s will may be changed until the end-of-life. In Japan, the percentage of patients undergoing TIV is higher than that in other countries, and in some cases, a high QOL is achieved even under TIV; therefore, measures that suit the Japanese background are required.
Communication support
Communication is paramount for maintaining patient autonomy and dignity. Ensuring communication through augmentative and alternative communication (AAC) enhances patient autonomy and improves the QOL of the patient and caregiver (Box 5). Multidisciplinary care is important because it is sometimes unacceptable to introduce the next stage of methods ahead of time, while functions remain. Every 6 months, the site(s) of voluntary movement should be re-evaluated for any incompatibility with the switch and adjusted for the input switch so that communication is maintained1). Social resources related to AAC support include those for physical support of the equipment itself and those for human support to implement and maintain AAC tools through adaptation, coordination, and guidance.
Box 5 Augmentative and alternative communication useful for people with amyotrophic lateral sclerosis.
Technology free
(without tools/IT) |
• kuchimoji (in Japanese), which comprised an oral shape and eye-blink method without a board |
| • signals (blinking etc.), hand writing |
| Alternative technology |
|
Low-tech
(tools without IT) |
flip chart and symbols (communication board of Japanese letters) simple keyboard for spelling of words |
| Boogie Board |
High-tech
(device using IT) |
general tablet |
| enhanced tablet to produce synthesized voice |
| bespoke tablet to produce synthesized voice |
| voice output communication aids |
| Japanese scanning communication aids |
| character input methods for eye-gaze input interface |
| novel interface that utilizes biological information or brain |
| machine interface |
IT, information technology.
Patient care support varies across countries and insurance systems; however, it is important to effectively use government support in collaboration with multidisciplinary staff.
In Japan, multiple systems such as medical insurance, long-term care insurance, disability welfare services, policies for intractable diseases, and informal services that have not been institutionalized are available to support patients with ALS in their medical care. Additionally, assistive and prosthetic devices are available for the treatment of daily life disabilities through long-term care insurance and disability welfare services. Rehabilitation is provided through medical and long-term care insurance. For respiratory disorders, medical insurance allows the use of expectorant support devices when patients are eligible for home ventilatory therapy instructions and management fees. Additionally, there is a support system called “respite hospitalization,” in which patients are temporarily hospitalized in specifically approved wards such as community-based integrated care units or based on policies for intractable diseases, to reduce the burden on caregivers and continue home care.
Disaster (emergency) preparedness
Patients and caregivers should prepare for disasters by making their homes earthquake-proof, preparing water and food, sourcing necessary medical and nursing supplies, and securing emergency power sources. Medical equipment that requires a power source, such as ventilators and suction devices, should be equipped with internal batteries, and their operating times should be confirmed (Table 7).
Table 7 Recommendations in instructions and support for people living with amyotrophic lateral sclerosis.
| Subject |
Recommendations |
| Multidisciplinary care |
• As patients with ALS often have difficulty with travelling, multidisciplinary care should be completed in a single periodic visit. |
| • Patients who receive multidisciplinary care may have longer survival, better quality of life, and fewer unplanned hospitalizations owing to pneumonia than those who do not receive multidisciplinary care. |
| Communicating the diagnosis |
• Inform the patient about his/her medical condition through multidisciplinary care. |
| • If ALS is likely to be confirmed, explain the current perception of the disease to the patient. |
| • If ALS cannot be ruled out, inform the patient that the diagnosis is uncertain but suspected. |
| — Explanation of heredity |
| • If there is no family history or consanguineous marriages, explain that inheritance is unlikely. |
| • If a family history exists and the patient requests information, explain the genetics of ALS. |
| • Take ethical considerations, such as genetic counseling, into account for genetic testing. |
| — Explanation for patients with dementia or depression |
| • When the patient has dementia, explain the condition to the family /key person and primary caregiver. |
| • For patients with depression, consult with the family/key person and primary caregiver to determine the best way to explain the patient’s medical condition, based on psychiatrist’s opinion. |
| • If the patient’s decision-making capacity is impaired, consult family members who understand the patient’s thinking and decide on the best approach. |
| Prognostic indicator of survival |
• Emphasize that weight maintenance and NIV improve prognosis. |
| • Simulating and communicating the means of communication, caregiving, and financial issues that may arise if TIV is introduced in the future are important. |
| • Medical treatment for disease progression should be explained as early as possible after diagnosis; time should be spent to understand the patient’s living and care environment. |
| • Gastrostomy for patients with impaired respiratory function should be performed under NIV. |
| • Surgery requiring general anesthesia should be avoided in patients with ALS. |
| • Even in cases where respiratory function is not impaired, careful attention should be paid to respiratory function, the degree of progression, and prognosis as well as whether the patient wishes to undergo TIV. |
| • Spondylolisthesis surgery in patients with ALS is unlikely to lead to improvement and has not been reported to prolong survival. |
| Shared decision-making (SDM) |
• A multidisciplinary care team is required to stay one step ahead but not plan too far ahead of the patient’s needs; staging of the provision of information and timing of support is important and should be carefully negotiated. |
| • SDM is a step-by-step process as follows: (1) Participant engagement, (2) Option information, (3) Option deliberation, (4) Decision implementation. |
| End-of-life care issues |
• Communicate what could happen at the end-of-life, for example, how death may occur. |
| • Advance care planning, including Advance Decisions to Refuse Treatment and Do Not Attempt resuscitation orders, should be communicated. |
| • Ensure that advanced care plans will be available when needed, for example, include the information on the patient’s Summary Care Record. |
| • Assuming possible symptoms, appropriate symptomatic treatment and care should be provided to maximize quality of life. For this purpose, the Palliative Care Scale (Box 6) is useful. |
| Communication support |
• AAC includes tools and technology-free measures (without tools/IT) and tools and technology measures (low-tech aids and high-tech aids) (Box 5). |
| • To promote AAC support, start while motor and speech-language functions remain, and try various methods during sufficient communication still maintained with an IT-free AAC (technology free or low-tech aid). |
| • Some patients leave their original voices while the functions remain to synthesize their own voices using new technologies. |
| • If it is difficult to detect involuntary movement, a biopotential signal sensor attached to the skin surface using Cybernics technology and switch operation from biological information such as electroencephalography signals or cerebral blood flow changes by means of a near-infrared spectroscopy will be attempted. |
| Care support |
Since support for patients’ medical care varies across countries and insurance systems, effective use of administrative support in cooperation with multidisciplinary staff is important. |
| Disaster (emergency) preparedness |
• Patients should prepare for disasters by making their homes earthquake-proof, preparing water and food, sourcing necessary medical and nursing supplies, and securing an emergency power supply. |
| • Patients and caregivers should discuss and confirm their safety, evacuation methods, evacuation routes, and evacuation sites in the event of a disaster. |
| • Medical equipment that requires a power source, such as ventilators and suction devices, should be equipped with built-in batteries, and their operating times should be confirmed. |
| • Prepare alternative equipment that does not require a power source, and understand the considerations for using a plug-in hybrid vehicle or generator as power source. |
ALS, amyotrophic lateral sclerosis; AAC, augmentative and alternative communication; IT, information technology; NIV, noninvasive ventilation; SDM, shared decision-making; TIV, tracheostomy invasive ventilation.
Box 6 Amyotrophic lateral sclerosis Palliative Care Scale assessed by patient-reported suffering
69).
| Symptoms or sufferings |
|
| Dyspnea |
Shortness of breath, difficulty breathing, difficulty breathing when moving |
| Pain |
Painful, aching joints, sore skin, tight muscles |
| Restlessness |
Inability to sit still, body not positioned, itching |
| Thirst |
Thirsty, dry mouth, need cold water |
| Burning sensation |
Body hot, head hot, need cool air |
| Choking |
Saliva and phlegm sticking to the throat, choking, coughing |
| Nausea |
Nausea, queasiness |
| Constipation |
Inability to pass stools, distended abdomen, and pain |
| Insomnia |
Inability to sleep, waking up several times, waking up early in the morning |
| Drooling |
Saliva leaks from the mouth |
| Fatigue |
Lack of energy, tiredness |
| Anxiety |
I am anxious about the future, I feel blocked, and I have worries. |
| Loneliness |
Lonely, hard to be alone, no one understands |
| Irritation |
Irritability, restlessness, and the desire to blame others |
| Communication difficulty |
I cannot get my ideas across to others, or they do not understand me. |
Methods
Guideline panel composition
The working group responsible for the development of these guidelines comprised 33 Japanese members. This included 13 core members (authors) led by M.A., 19 collaborators, and one external reviewer. The core members comprised 12 neurologists (all authors except Y.N.) and one nurse (Y.N.). To ensure unbiased recommendations, an independent panel of 10 Japanese members was established. The panel comprised neurologists, nurses, medical social workers, and patients with ALS. Notably, five members (M.A., T.S., H.W., M.M., and K.N.) had previously participated in the working group. Four members among the collaborators (N.O., A.K., Y.O., and S.H.) were independently involved in analyzing the SR and summarizing the evidence for decision-making purposes.
Literature search
In November 2019, a preparatory committee was established to revise the 2013 ALS Clinical Practice Guidelines as much as possible in accordance with the Minds Clinical Practice Guideline Preparation Manual 2017 and 2020 ver. 3.0 by the Japan Council for Quality Health Care. On January 31, 2020, the Board of Directors of the JSN approved the ALS Clinical Practice Guidelines Development Committee. The first drafting committee meeting was held on February 22, 2020. Owing to the coronavirus disease 2019 pandemic, the remaining meetings were held online (final date: February 11, 2022). To scrutinize the current state of evidence, the Japan Medical Library Association conducted an unbiased literature search of English and Japanese papers published between 2000 and 2020 using three databases: the Cochrane Library, PubMed, and Ichushi-Web. After the search, other essential documents and secondary reference materials obtained by December 31, 2021, were added by committee members. The search strategies are available in the appendix of the full guidelines (www.neurology-jp.org/guidelinem/, in Japanese).
Review process
Two questions with sufficient evidence were identified to make the recommendation: (1) Is riluzole recommended for patients with ALS? (2) Is edaravone recommended for patients with ALS? NIV, which is widely used in clinical practice, was also discussed but was included in the expert consensus because of the low level of evidence (only one RCT).
The certainty of evidence for each outcome was assessed based on study design, risk of bias, inconsistency, indirectness, and imprecision. The GRADE evidence profile and qualitative SR of each outcome were summarized as research evidence in the evidence-to-decision framework, according to the following criteria: (1) Is the problem a priority? (2) How substantial are the anticipated desirable effects? (3) How substantial are the anticipated undesirable effects? (4) What is the overall certainty of evidence for these effects? (5) Does the balance between desirable and undesirable effects favor intervention or comparison? (6) Is the intervention acceptable to key stakeholders? (7) Is the intervention feasible for patients, their caregivers, and healthcare providers?
Based on the evidence-to-decision framework, we developed an executive summary of the guideline statements and a working group, composed of a Guideline Development Committee and an Independent Panel, and judged the acceptability and appropriateness of each criterion based on the research evidence. Subsequently, the working group voted on the strength of the recommendation (Box 7) and summarized the body of evidence (Box 8). Voting was conducted via the web (Google form®) with a fixed voting period. To determine the level of recommendation, the following two points were considered in advance: (a) If > 50% of the committee members were in favor of the intervention and < 20% of the committee members considered the comparison intervention “strongly preferable,” a recommendation in favor of the intervention was selected. (b) Additionally, if > 70% support the “strong recommendation,” the “strong recommendation” was adopted
Box 7 Strength of recommendation.
| • Strong recommendation for using an intervention |
| • Weak recommendation for using an intervention |
| • Weak recommendation against using an intervention |
| • Strong recommendation against using an intervention |
| • Abstention due to conflict of interest |
Box 8 Strength of evidence.
| A (strong) |
The panel is confident that the true effect lies close to that of the estimate of the effect to support the recommendation. |
| B (moderate) |
The panel is moderately confident in the effect estimate to support the recommendation. |
| C (weak) |
The panel’s confidence in the effect estimate is limited to support the recommendation. |
| D (very weak) |
The panel has extremely low confidence in the effect estimate to support the recommendation. |
The chairman, members, research collaborators, SR members, clinical question independent panel committee members, evaluation and coordinating members, and external members of the Guideline Development Committee reported individual conflicts of interest (COI) to the COI Committee of the JSN. The JSN manages COI based on the “Common Guidelines for Conflict of Interest in Medical Research.” Eight members met the COI disclosure standards of the JSN. This guideline was fully funded by the JSN. The JSN provided the actual expenses related to the conference held by the secretariat and a video platform for online conferences. Members of the Guideline Development Committee only paid transportation expenses to participate in the conference.
Concluding remarks
In conclusion, ALS remains an intractable disease with no cure; however, the course, prognosis, and QOL of patients can be improved through various treatments, interventions, and support. These guidelines reflect the current situation in Japan and may not be universally applicable to all patients with ALS. They are not intended to limit the discretion of medical practitioners. In the realm of personalized medicine, collaborative decision-making is imperative for assessing the patient’s condition, considering the values of both the patient and caregivers and accommodating cultural, regional, and social diversity.
Of particular significance is the provision of appropriate support and care, including invasive measures such as mechanical ventilation with tracheostomy, for both patients undergoing the procedure and those not undergoing it. Further research and advancements in this area are necessary, and this guideline has been developed with these considerations in mind, with a commitment to continuous improvement.
Additionally, the knowledge gap between Japan and other countries needs to be acknowledged. For instance, in the United States, sodium phenylbutyrate-taurursodiol38) and tofelsen39) have been approved for ALS in addition to riluzole and edaravone. In particular, tofelsen is the first antisense oligonucleotide in ALS targeting mutant SOD1-associated ALS cases. Moreover, the approval of oral edaravone in Japan in December 2022 alleviated the inconvenience associated with daily intravenous injections of edaravone. The recommendations for new drugs will be updated in the future.
Finally, we believe that this article will help improve and ensure the quality of clinical practice for patients with ALS in medical care settings and will be a cornerstone for further progress.
Collaborators
Research collaborators: Kensuke Ikeda, Tamotsu Imura, Hina Uetake, Ryosuke Oki, Masaya Oda, Yuji Tanaka, Kenichiro Nakazawa, Tameto Naoi, Sonoko Nozaki, Yasushi Fujimoto, Satoshi Yamashita, Yasuhiro Watanabe, Nobuhiro Ogawa, Akihiro Kitamura, Takako Saotome, Michiko Nakai, Koji Fujita, Chiharu Matsuda, and Ryosuke Miyamoto
The SR committee members: Nobuhiro Ogawa, Akihiro Kitamura, Masaya Oda, Shotaro Haji, Koji Fujita, and Ryosuke Miyamoto
The members of Independent Panel Committee on CQ: Reiko Ishiyama, Naoko Ideguchi, Miho Iwaki, Mitsuko Ushikubo, Shigeyuki Shimamori, Tatsuki Sugisawa, Fumi Nakamoto, Michio Nonaka, Hirofumi Maruyama, and Koichiro Miho
Funding
This work was fully supported by JSN.
Disclosure
Companies and organizations with COI that should be disclosed related to this paper are as follows: Masashi Aoki (lecture fees: Otsuka Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., Mitsubishi Tanabe Pharma Corporation), Yuishin Izumi (research expenses: Takeda Pharmaceutical Co., Ltd.), Makoto Urushitani (lecture fee: Mitsubishi Tanabe Pharma Corporation; research expenses: Astellas Pharma Inc., KAN Research Institute, Inc.; scholarship donation: Ono Pharmaceutical Co., Ltd., Bayer Yakuhin Ltd.), and Osamu Kano (lecture fees: Chugai Pharmaceutical Co., Ltd.; Scholarship Donation: Eisai Co., Ltd., Otsuka Pharmaceutical Co., Ltd.). Otherwise, there is no conflict of interest to declare.
Acknowledgments
The authors are grateful to Yoshiharu Ikeda, Katsuo Shimizu, and Ayano Fukano from the secretariat of the Japanese Society of Neurology for their generous support. As the external reviewers, Tateo Nakayama and Hidekazu Tomimoto on behalf of the Japanese Society of Neurological Therapeutics provided critical feedback with regard to the description of the guideline. We thank Editage (www.editage.jp) for English language editing. The Research Committee of CNS Degenerative Diseases (20FC1049 and 23FC1008), Research on Policy Planning and Evaluation for Rare and Intractable Diseases, Health, Labour and Welfare Sciences Research Grants, the Ministry of Health, Labour and Welfare, Japan is also acknowledged for their generous support. Finally, we would like to thank Akiko Okumura and Toshio Morizane from the Japan Council for Quality Health Care, and Shin-ichi Abe from the Japan Medical Library Association.
References
- 1) Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009;73:1227-1233.
- 2) EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis; Andersen PM, Abrahams S, Borasio GD, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)—revised report of an EFNS task force. Eur J Neurol 2012;19:360-375.
- 3) Motor Neurone Disease: Assessment and Management. London; 2019.
- 4) Boostani R, Olfati N, Shamshiri H, et al. Iranian clinical practice guideline for amyotrophic lateral sclerosis. Front Neurol 2023;14:1154579.
- 5) Petri S, Grehl T, Grosskreutz J, et al. Guideline “Motor neuron diseases” of the German Society of Neurology (Deutsche Gesellschaft fur Neurologie). Neurol Res Pract 2023;5:25.
- 6) Shoesmith C, Abrahao A, Benstead T, et al. Canadian best practice recommendations for the management of amyotrophic lateral sclerosis. CMAJ 2020;192:E1453-E1468.
- 7) Doi Y, Atsuta N, Sobue G, et al. Prevalence and incidence of amyotrophic lateral sclerosis in Japan. J Epidemiol 2014;24:494-499.
- 8) Nakamura R, Sone J, Atsuta N, et al. Next-generation sequencing of 28 ALS-related genes in a Japanese ALS cohort. Neurobiol Aging 2016;39:219.e1-219.e8.
- 9) Nishiyama A, Niihori T, Warita H, et al. Comprehensive targeted next-generation sequencing in Japanese familial amyotrophic lateral sclerosis. Neurobiol Aging 2017;53:194.e1-194.e8.
- 10) Mejzini R, Flynn LL, Pitout IL, et al. ALS genetics, mechanisms, and therapeutics: where are we now? Front Neurosci 2019;13:1310.
- 11) Iida A, Takahashi A, Kubo M, et al. A functional variant in ZNF512B is associated with susceptibility to amyotrophic lateral sclerosis in Japanese. Hum Mol Genet 2011;20:3684-3692.
- 12) Mathis S, Goizet C, Soulages A, et al. Genetics of amyotrophic lateral sclerosis: a review. J Neurol Sci 2019;399:217-226.
- 13) Fujimura-Kiyono C, Kimura F, Ishida S, et al. Onset and spreading patterns of lower motor neuron involvements predict survival in sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2011;82:1244-1249.
- 14) Turner MR, Barohn RJ, Corcia P, et al. Primary lateral sclerosis: consensus diagnostic criteria. J Neurol Neurosurg Psychiatry 2020;91:373-377.
- 15) Watanabe H, Atsuta N, Nakamura R, et al. Factors affecting longitudinal functional decline and survival in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener 2015;16:230-236.
- 16) Hayashi N, Atsuta N, Yokoi D, et al. Prognosis of amyotrophic lateral sclerosis patients undergoing tracheostomy invasive ventilation therapy in Japan. J Neurol Neurosurg Psychiatry 2020;91:285-290.
- 17) Nakayama Y, Shimizu T, Mochizuki Y, et al. Predictors of impaired communication in amyotrophic lateral sclerosis patients with tracheostomy-invasive ventilation. Amyotroph Lateral Scler Frontotemporal Degener 2015;17:38-46.
- 18) Nakayama Y, Shimizu T, Matsuda C, et al. Non-motor manifestations in ALS patients with tracheostomy and invasive ventilation. Muscle Nerve 2018;57:735-741.
- 19) Feldman EL, Goutman SA, Petri S, et al. Amyotrophic lateral sclerosis. Lancet 2022;400:1363-1380.
- 20) Clemens KE, Klaschik E. Morphine in the management of dyspnoea in ALS. A pilot study. Eur J Neurol 2008;15:445-450.
- 21) Oliver DJ, Campbell C, O'Brien T, et al. Medication in the last days of life for motor neuron disease/amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2010;11:562-564.
- 22) Bello-Haas VD. Physical therapy for individuals with amyotrophic lateral sclerosis: current insights. Degener Neurol Neuromuscul Dis 2018;8:45-54.
- 23) Majmudar S, Wu J, Paganoni S. Rehabilitation in amyotrophic lateral sclerosis: why it matters. Muscle Nerve 2014;50:4-13.
- 24) Janse van Mantgem MR, van Eijk RPA, van der Burgh HK, et al. Prognostic value of weight loss in patients with amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry 2020;91:867-875.
- 25) Steyn FJ, Ioannides ZA, van Eijk RPA, et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J Neurol Neurosurg Psychiatry 2018;89:1016-1023.
- 26) Ludolph AC, Dorst J, Dreyhaupt J, et al. Effect of high-caloric nutrition on survival in amyotrophic lateral sclerosis. Ann Neurol 2020;87:206-216.
- 27) Kurihara M, Bamba S, Yasuhara S, et al. Factors affecting energy metabolism and prognosis in patients with amyotrophic lateral sclerosis. Ann Nutr Metab 2021;77:236-243.
- 28) Shimizu T, Ishikawa-Takata K, Sakata A, et al. The measurement and estimation of total energy expenditure in Japanese patients with ALS: a doubly labelled water method study. Amyotroph Lateral Scler Frontotemporal Degener 2017;18:37-45.
- 29) Fasano A, Fini N, Ferraro D, et al. Percutaneous endoscopic gastrostomy, body weight loss and survival in amyotrophic lateral sclerosis: a population-based registry study. Amyotroph Lateral Scler Frontotemporal Degener 2017;18:233-242.
- 30) McDonnell E, Schoenfeld D, Paganoni S, et al. Causal inference methods to study gastric tube use in amyotrophic lateral sclerosis. Neurology 2017;89:1483-1489.
- 31) Ichihara N, Namba K, Ishikawa-Takata K, et al. Energy requirement assessed by doubly-labeled water method in patients with advanced amyotrophic lateral sclerosis managed by tracheotomy positive pressure ventilation. Amyotroph Lateral Scler 2012;13:544-549.
- 32) Aoun SM, Breen LJ, Edis R, et al. Breaking the news of a diagnosis of motor neurone disease: A national survey of neurologists’ perspectives. J Neurol Sci 2016;367:368-374.
- 33) Hirayama T, Izumi Y, Nakayama Y, et al. Communicating the diagnosis: a survey of patients with amyotrophic lateral sclerosis and their families in Japan. Acta Neurol Belg 2022;122:471-478.
- 34) McCluskey L, Casarett D, Siderowf A. Breaking the news: a survey of ALS patients and their caregivers. Amyotroph Lateral Scler Other Motor Neuron Disord 2004;5:131-135.
- 35) Hogden A, Foley G, Henderson RD, et al. Amyotrophic lateral sclerosis: improving care with a multidisciplinary approach. J Multidiscip Healthc 2017;10:205-215.
- 36) Yoshor D, Klugh A, 3rd, Appel SH, et al. Incidence and characteristics of spinal decompression surgery after the onset of symptoms of amyotrophic lateral sclerosis. Neurosurgery 2005;57:984-989; discussion 984-989.
- 37) Sancho J, Servera E, Chiner E, et al. Noninvasive respiratory muscle aids during PEG placement in ALS patients with severe ventilatory impairment. J Neurol Sci 2010;297:55-59.
- 38) Paganoni S, Macklin EA, Hendrix S, et al. Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N Engl J Med 2020;383:919-930.
- 39) Miller TM, Cudkowicz ME, Genge A, et al. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med 2022;387:1099-1110.
- 40) Geevasinga N, Loy CT, Menon P, et al. Awaji criteria improves the diagnostic sensitivity in amyotrophic lateral sclerosis: a systematic review using individual patient data. Clin Neurophysiol 2016;127:2684-2691.
- 41) Shefner JM, Al-Chalabi A, Baker MR, et al. A proposal for new diagnostic criteria for ALS. Clin Neurophysiol 2020;131:1975-1978.
- 42) Watanabe Y, Raaphorst J, Izumi Y, et al. Cognitive and behavioral status in Japanese ALS patients: a multicenter study. J Neurol 2020;267:1321-1330.
- 43) Carvalho TL, de Almeida LM, Lorega CM, et al. Depression and anxiety in individuals with amyotrophic lateral sclerosis: a systematic review. Trends Psychiatry Psychother 2016;38:1-5.
- 44) Thakore NJ, Pioro EP. Laughter, crying and sadness in ALS. J Neurol Neurosurg Psychiatry 2017;88:825-831.
- 45) Boentert M. Sleep disturbances in patients with amyotrophic lateral sclerosis: current perspectives. Nat Sci Sleep 2019;11:97-111.
- 46) Jackson CE, McVey AL, Rudnicki S, et al. Symptom management and end-of-life care in amyotrophic lateral sclerosis. Neurol Clin 2015;33:889-908.
- 47) Vogt S, Schreiber S, Pfau G, et al. Dyspnea as a fatigue-promoting factor in ALS and the role of objective indicators of respiratory impairment. J Pain Symptom Manage 2020;60:430-438.e1.
- 48) Chiò A, Mora G, Lauria G. Pain in amyotrophic lateral sclerosis. Lancet Neurol 2017;16:144-157.
- 49) Hobson EV, McDermott CJ. Supportive and symptomatic management of amyotrophic lateral sclerosis. Nat Rev Neurol 2016;12:526-538.
- 50) Andrews JA, Meng L, Kulke SF, et al. Association between decline in slow vital capacity and respiratory insufficiency, use of assisted ventilation, tracheostomy, or death in patients with amyotrophic lateral sclerosis. JAMA Neurol 2018;75:58-64.
- 51) Chatwin M, Toussaint M, Goncalves MR, et al. Airway clearance techniques in neuromuscular disorders: a state of the art review. Respir Med 2018;136:98-110.
- 52) Bourke SC, Tomlinson M, Williams TL, et al. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol 2006;5:140-147.
- 53) Dorst J, Ludolph AC. Non-invasive ventilation in amyotrophic lateral sclerosis. Ther Adv Neurol Disord 2019;12:1756286419857040.
- 54) Morelot-Panzini C, Bruneteau G, Gonzalez-Bermejo J. NIV in amyotrophic lateral sclerosis: The ‘when’ and ‘how’ of the matter. Respirology 2019;24:521-530.
- 55) Park D, Lee GJ, Kim HY, et al. Different characteristics of ventilator application between tracheostomy- and noninvasive positive pressure ventilation patients with amyotrophic lateral sclerosis. Medicine (Baltimore) 2017;96:e6251.
- 56) Vianello A, Arcaro G, Molena B, et al. Effect of a passive exhalation port on tracheostomy ventilation in amyotrophic lateral sclerosis patients: a randomized controlled trial. J Thorac Dis 2018;10:1007-1014.
- 57) Krivickas LS, Dal Bello-Hass V, Danforth SE, et al. Rehabilitation. New York: Taylor and Francis Group; 2006.
- 58) Nakajima T, Sankai Y, Takata S, et al. Cybernic treatment with wearable cyborg Hybrid Assistive Limb (HAL) improves ambulatory function in patients with slowly progressive rare neuromuscular diseases: a multicentre, randomised, controlled crossover trial for efficacy and safety (NCY-3001). Orphanet J Rare Dis 2021;16:304.
- 59) Hanson EK. Dysarthria in amyotrophic lateral sclerosis: a systematic review of characteristics, speech treatment, and augmentative and alternative communication. J Med Speech Lang Pathol 2011;19:12-30.
- 60) Tomik B, Guiloff RJ. Dysarthria in amyotrophic lateral sclerosis: a review. Amyotroph Lateral Scler 2010;11:4-15.
- 61) Connors K, Mahony L, Morgan P. Variation in assistive technology use in motor neuron disease according to clinical phenotypes and ALS functional rating scale—revised score: a prospective observational study. NeuroRehabilitation 2019;44:303-313.
- 62) Soofi AY, Bello-Haas VD, Kho ME, et al. The impact of rehabilitative interventions on quality of life: a qualitative evidence synthesis of personal experiences of individuals with amyotrophic lateral sclerosis. Qual Life Res 2018;27:845-856.
- 63) Bourke SC, Bullock RE, Williams TL, et al. Noninvasive ventilation in ALS: indications and effect on quality of life. Neurology 2003;61:171-177.
- 64) Traynor BJ, Alexander M, Corr B, et al. Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study, 1996-2000. J Neurol Neurosurg Psychiatry 2003;74:1258-1261.
- 65) Galvin M, Corr B, Madden C, et al. Caregiving in ALS—a mixed methods approach to the study of Burden. BMC Palliat Care 2016;15:81.
- 66) Poletti B, Carelli L, Solca F, et al. Sexuality and intimacy in ALS: systematic literature review and future perspectives. J Neurol Neurosurg Psychiatry 2019;90:712-719.
- 67) Ingre C, Chen L, Zhan Y, et al. Lipids, apolipoproteins, and prognosis of amyotrophic lateral sclerosis. Neurology 2020;94:e1835-e1844.
- 68) Baile WF, Buckman R, Lenzi R, et al. SPIKES—a six-step protocol for delivering bad news: application to the patient with cancer. Oncologist 2000;5:302-311.
- 69) Shimizu T, Shimizu N, Onozaki K, et al. A proposal of a novel palliative care scale and analysis of suffering in amyotrophic lateral sclerosis. Rinsho Shinkeigaku 2021;61:361-367.
Abbreviations/Glossary
AACaugmentative and alternative communication
ALSamyotrophic lateral sclerosis
ALSFRS-RALS functional scale-revised
COIConflict of Interest
CQclinical question
EtDevidence-to-decision
FALSfamilial ALS
GRADE, grading of recommendations assessmentdevelopment and evaluation
JSNthe Japanese Society of Neurology
MRImagnetic resonance imaging
NIVnon-invasive ventilation
PEGpercutaneous endoscopic gastrostomy
PICO, population, intervention, comparisonand outcomes
PLSprimary lateral sclerosis
PMAprogressive muscular atrophy
QOLquality of life
RCTrandomized control trial
SDMshared decision making
SRsystematic review
TIVtracheostomy invasive ventilation
