Prdm12 Is Induced by Retinoic Acid and Exhibits Anti-proliferative Properties through the Cell Cycle Modulation of P19 Embryonic Carcinoma Cells
2013 年 38 巻 2 号 p. 197-206

詳細
2013 年 38 巻 2 号 p. 197-206
The Prdm (PRDI-BF1-RIZ1 homologous) family is involved in cell differentiation, and several Prdms have been reported to methylate histone H3 by intrinsic or extrinsic pathways. Here, we report that Prdm12 recruits G9a to methylate histone H3 on lysine 9 through its zinc finger domains. Because of the expression of Prdm12 in the developmental nervous system, we investigated the role of Prdm12 on P19 embryonic carcinoma cells as a model system for neurogenesis. In P19 cells, Prdm12 is induced by Retinoic acid (RA). Overproduction of Prdm12 in P19 cells impairs cell proliferation and increases the G1 population accompanied by the upregulation of p27. In contrast, the knockdown of Prdm12 increases the number of cells in a suspension culture of RA-induced neural differentiation. Both the PR domain and zinc finger domains are required for the anti-proliferative activity of Prdm12. While the data in this study is based on in vitro models, the results suggest that Prdm12 is induced by the RA signaling in vivo, and may regulate neural differentiation during animal development.
The Prdm family contains a PR (PRDI-BF1-RIZ1 homologous) domain in the N-terminal which has a 20–30% sequence identity to the conserved catalytic SET (SU(VAR) 3–9, E(Z), trithorax) domains of the histone lysine methyltransferase (HKMTase) superfamily (Völkel and Angrand, 2007). This similarity suggests that the Prdm family may possess HKMTase properties. In fact, some Prdms show intrinsic HKMTase activity (Prdm2, Prdm3, Prdm8, Prdm9, and Prdm16). In addition, Prdm1, Prdm5, and Prdm6 lack intrinsic HKMTase activity, but instead recruit G9a/Ehmt2/KMT1C, a strong mammalian histone H3 lysine 9 (H3K9) methyltransferase, to mediate HKMTase activity (see Fog et al., 2012 for a review). Another structural feature is that the Prdm family has multiple kruppel-type zinc finger (ZF) domains in the C-terminus involved in sequence-specific DNA binding and protein-protein interactions (Völkel and Angrand, 2007).
It has been reported that the Prdm family is expressed dynamically in the nervous system during the development of mice and zebrafish (Hohenauer and Moore, 2012). The function of the Prdms in neurogenesis remains unclear, however. In zebrafish embryos, Prdm1 is important for the development of neural crest and sensory neurons (Hernandez-Lagunas et al., 2005). Our laboratory has previously described the expression pattern of Prdm8 in the mouse embryonic nervous system (Komai et al., 2009). Another paper using a knockout mice model indicated that Prdm8 is involved in neural development (Ross et al., 2012). Although Prdm12 is expressed in the mouse and zebrafish embryonic nervous system (Kinameri et al., 2008; Sun et al., 2008), the role of Prdm12 remains unknown.
Retinoic acid (RA) is a metabolite of vitamin A (retinol), which controls neural differentiation and patterning (Maden, 2007). To investigate the process of neural differentiation in vitro, P19 embryonic carcinoma cells were used as a model system. Treatment with RA in aggregating condition induces P19 cells to differentiate into neurons and glia (Jones-Villeneuve et al., 1982). The upregulation of p27, a cyclin-dependent kinase inhibitor (CKI), has been shown to be involved in arresting cell cycle progression at G1 phase in this in vitro model (Gill et al., 1998). Furthermore, the overexpression of p27 could induce neural differentiation in mouse neuroblastoma cells (Kranenburg et al., 1995).
Since many Prdms possess HKMTase properties, we are interested in whether Prdm12 also possesses HKMTase properties. In this study, we first demonstrate that Prdm12 recruits G9a to methylate H3K9. Because the localization in the embryonic nervous system implies a potential function in neurogenesis, we investigated the role of Prdm12 in the RA-induced neural differentiation of P19 cells. We discover that Prdm12 is induced by RA in P19 cells and exhibits anti-proliferative effects partially through the regulation of G1 phase in the cell cycle. Furthermore, ectopic Prdm12 increases the expression of p27 with both the PR and ZF domains being necessary for its function.
Full-length Prdm12 cDNA was amplified by PCR from an EST clone (image #6734548) purchased from the IMAGE consortium. For the generation of expression plasmids, amplified Prdm12 cDNA was inserted into pEGFP-C2 (Clontech, Mountair View, CA, USA), pGEX4T-1 (GE Healthcare, Little Chalfont, UK), or pCAG-FLAG-IRES-Puro vectors at specific restrict enzyme sites in frame with the indicated tag. To make Prdm12 deletion and point mutants, mutated fragments were created by PCR amplification and were subcloned into a pCAG-FLAG-IRES-Puro vector. Glutathione S-transferase (GST)-tagged H3N, mutants, and GST-G9aSET have been described earlier (Tachibana et al. 2001). Three oligonucleotides of short hairpin RNAs (shRNA) targeting the Prdm12 gene were designed and synthesized from Sigma (St. Louis, MO, USA) then inserted into a pSUPER.retro.puro vector (OligoEngine, Seattle, WA). Details for all the primers and restriction enzyme sites are listed in the supplemental information (Table. SI).
Cell culture and differentiationHEK293T, NIH3T3, and P19 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Nissui Pharmaceutical Co. Ltd.) supplemented with 10% fetal bovine serum (FBS), 100 μg/ml streptomycin, and 100 U/ml penicillin (Gibco, Carlsbad, CA, USA) at 37°C in a 5% CO2 atmosphere. To induce neural differentiation, 1×106 P19 cells were cultured on 10 cm bacteria grade dishes for aggregation in DMEM supplemented with 10% FBS and 1 μM RA (solved in 99.9% EtOH; Sigma). After 96 h, cells were trypsinized then transferred to poly-L-lysine (Sigma) coated tissue culture dishes at a density of 1×105 cells/ml in 10% FBS DMEM without RA. After 24 h, the medium was changed to 0.5% FBS DMEM to induce neural differentiation for another 96 h. All media were replaced with fresh media every 48 h.
Transfection and infection of cellsHEK293T, NIH3T3, and P19 cells were seeded in 6-well culture plate for 24 h before transfection. Two micrograms of indicated plasmids were transfected into HEK293T cells using TransIT-LT-1 reagent (Mirus Bio corp., Madison, WI, USA) or 4 μg of plasmids were transfected into NIH3T3 and P19 cells using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. For infection, GP2–293 cells were co-transfected with a pSUPER.retro.puro vector containing the shRNA against mouse Prdm12 and a pVSV-G vector. After 48 h, the culture media was filtered with a 0.45-μm filter then used to infect P19 cells with polybrene (8 μg/ml). To establish stable cell lines, cells were treated with 1 μg/ml puromycin for five days.
In vitro histone methyltransferase (HMTase) assayHMTase assays was performed as described (Tachibana et al. 2001), with some modifications. Briefly, 10 μl of reaction mixture containing 2 μg of core histones, immunoprecipitated enzymes, and 125 nCi of S-adenosyl-[methyl-14C]-L-methionine in assay buffer (50 mM Tris, pH 8.5, 5 mM DTT) were incubated for 1 h at 30°C. Proteins were separated by 15% SDS-PAGE and visualized by coomassie brilliant blue staining. Detection of methyl-14C was performed by using a BAS-5000 imaging analyzer (Fuji Film).
Immunoprecipitation and immunocytochemistryAfter 48 h of transfection (or at the harvesting times indicated), cells were lysed in lysis buffer containing 20 mM Hepes, pH 7.5, 420 mM NaCl, 1.5 mM MgCl2, 0.1% NP-40, and protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan). Supernatants were collected by centrifugation then incubated with 0.5–1 μg antibody overnight at 4°C and subsequently isolated by protein G-agarose (GE Healthcare). Immunoprecipitants were washed extensively with lysis buffer then used in western blot or in vitro HMTase assays. For immunocytochemistry, cells were fixed and immunostained as previous described (Komai et al. 2009).
AntibodiesTo generate rabbit anti-Prdm12 polyclonal antibodies, cDNA fragments corresponding to residues N43–F230 of mouse Prdm12 were subcloned into pGEX-4T-1, then the fusion protein was purified and injected into a rabbit (Hokudo Co., Ltd., Sapporo, Japan). The antibody was affinity-purified from the antiserum. The commercially available primary antibodies used were as follows: mouse monoclonal antibodies to FLAG M2 (F3165; Sigma); GFP (11 814 460 001; Roche, Mannheim, Germany); G9a (A8620A, Perseus Proteomics Inc., Tokyo, Japan); GLP (#422, Perseus Proteomics Inc.); Neuronal Class III β-Tubulin (Tuj1, MMS-435P; (Covance, Princeton, USA); p27 (K25020; BD Biosciences, San Jose, CA); α-tubulin (T-5168, Sigma) and normal rabbit IgG (sc-2027; Santa Cruz Biotechnology, Santa Cruz, CA); and rabbit polyclonal antibodies to p15 (#4822; Cell Signaling, Danvers, MA). Secondary antibodies were as follows: peroxidase-conjugated mouse or rabbit IgG secondary antibodies (NA931V and NA934V; GE Healthcare) and anti-mouse IgG Alexa 568 (A11004; Invitrogen).
Cell counting and cell cycle analysisFLAG-Prdm12 or mutants overexpressing P19 cells were seeded in 6 well plates at a density of 1×105 cells/well. Cell numbers were counted under a light microscope by trypan blue exclusion every 48 h for 6 days. The division times were calculated according to the following formula: (lgND–lgN0)/ lg2, where N0 is the cell number on the day of seeding, and ND is the cell number after culture for 2, 4, or 6 days. For cell cycle analysis, 1×106 FLAG-Prdm12 or mutants overexpressing P19 cells were seeded into 10-cm culture dishes. After 24 h, cells were collected, fixed, stained, and analyzed using flow cytometer as previously described (Tachibana et al. 2002).
Quantitative RT-PCRTotal RNAs were isolated with Sepasol-RNA I (Nacalai Tesque) on the indicated days after RA treatment and cDNA was synthesized using the Omniscript RT kit (QIAGEN, KJ Venlo, Nether-lands) according to the manufacturer’s instructions. Quantitative RT-PCR was performed with Power SYBR Green PCR Master Mix (Applied Biosystems, Warrington, UK) on a StepOne Plus Real-Time PCR System (Applied Biosystems). The primers used were Prdm12 forward 5′-CCCTTTTGGGGCTTCAGTTTC-3′, and reverse 5′-GGTCGCTCATTCTCTTGTTTGG-3′; G9a forward 5′-CACAAGCACATCGATGTGATT-3′, and reverse 5′-ATGGTAGTTGACAGCATGGAG-3′; Oct3/4 forward 5′-GAAGCAGAAGAGGATCACCTTG-3′, and reverse 5′-TTCTTAAGGCTGAGCTGCAAG-3′; Tuj1 forward 5′-TGGACAGTGTTCGGTCTGG-3′, and reverse 5′-CCTCCGTATAGTGCCCTTTGG-3′; GAPDH forward 5′-CATCTTCTTGTGCAGTGCCA-3′, and reverse 5′-CGTTGATGGCAACAATCTCC-3′. Gene expression was analyzed by the ΔΔ-CT method through StepOne Software 2.1 (Applied Bio-systems).
We hypothesized that Prdm12 may possess HKMTase properties. To investigate the potential HKMTase properties of Prdm12, we expressed and purified enhanced green fluorescent protein (EGFP)-tagged Prdm12 (EGFP-Prdm12), EGFP-tagged Prdm4, and EGFP from HEK293T cells for in vitro methylation assays using core histones as substrates. Glutathione S-transferase (GST)-fused catalytic SET domain of G9a (GST-G9aSET) was added as a positive control. The EGFP-Prdm12 showed higher H3 methylation than EGFP or EGFP-Prdm4 (Fig. 1A), suggesting that EGFP-Prdm12 can methylate histone H3. However, G9a is a strong H3K9 methytransferase; compared to G9a the HKMTase activity of Prdm12 is weak. Next, we determined which site on H3 was methylated by Prdm12. To this end, GST-fused histone H3 amino-terminal (residues 1–57, GST-H3N) and mutants in which single K substitutions were introduced to lysine 4, 9 or 27 (GST-H3NK4R, GST-H3NK9R, and GST-H3NK27R) were purified from Escherichia coli as substrates for the in vitro methylation assay (Fig. 1B). GST-H3N, GST-H3NK4R and GST-H3NK27R, but not GST-H3NK9R, were methylated by EGFP-Prdm12 (Fig. 1C). This result indicated that EGFP-Prdm12 specifically methylates lysine 9 of Histone 3 (H3K9). To further examine whether the HKMTase activity of Prdm12 was intrinsic or extrinsic, we expressed and purified GST-fused Prdm12 (GST-Prdm12) from E. coli for an in vitro methylation assay. However, we did not observe HKMTase activity in GST-Prdm12 (Fig. 1D). These results implied that Prdm12 may lack intrinsic HKMTase activity but instead forms complexes with other enzymes possessing H3K9 methyltransferase properties.
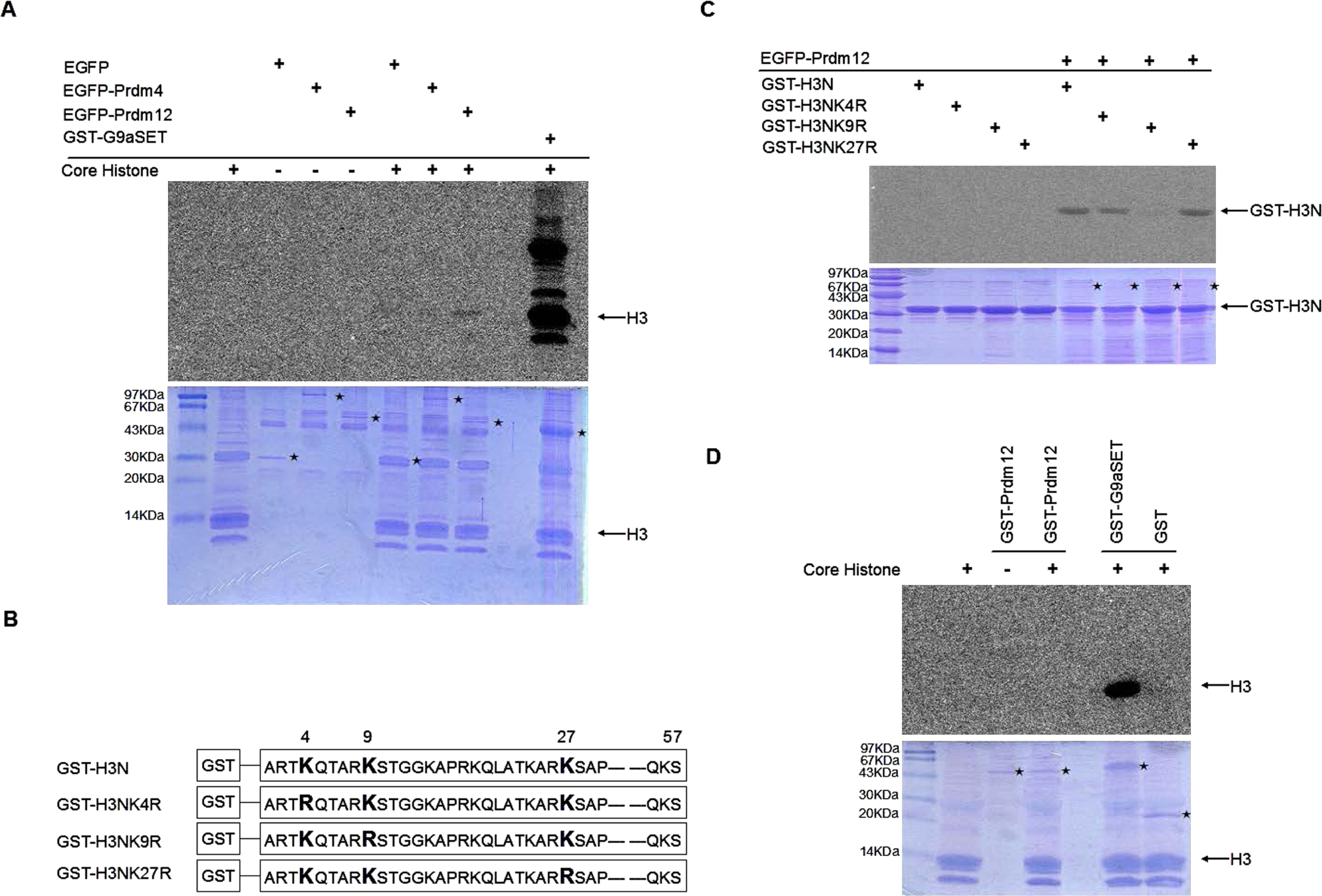
The Prdm12 complex mediates the methylation of histone H3 lysine in vitro. (A) In vitro methylation assay. EGFP-Prdm12, EGFP-Prdm4, or GFP plasmids were transiently transfected into HEK293T cells and purified by anti-GFP antibodies. GST-G9aSET, were expressed in E. coli and purified with glutathione-Sepharose beads as a positive control. The immunoprecipitated complex was incubated with 2 μg core histones as substrates and S-adenosyl-[methyl-C14]-L-methionine as a methyl donor. After incubation for 60 min at 30°C, samples were subjected to 15% SDS-PAGE. Methylated histones were detected by autoradiography (top). The amount of protein was shown by coomassie brilliant blue (bottom). ⋆, enzymes. (B) Schematic diagram of GST-H3N and point mutants (Tachibana et al. 2001). Residues 1–57 of the N-terminal of histone H3 were fused to GST. The lysine (K) was replaced by arginine (R) at the indicated position. (C) Two micrograms of GST-H3N or mutants were used as substrates for the in vitro methylation assay as described in (A). (D) GST-Prdm12, GST-G9aSET, and GST were expressed in E. coli and purified with glutathione-Sepharose beads. One microgram of each enzyme was incubated with 2 μg core histones as substrates for in vitro methylation assay as described in (A). Methylated histones were detected by autoradiography (top). The amount of protein was revealed by coomassie brilliant blue (bottom). ⋆, enzymes.
Prdm1, Prdm5, and Prdm6 were reported to mediate HKMTase activity by recruiting G9a. To examine whether Prdm12 could interact with G9a in a similar way to the Prdm proteins, we performed co-immunoprecipitation assays (Co-IP). EGFP-Prdm12 and FLAG-tagged G9a (FLAG-G9a) were transiently expressed in HEK293T cells. The IP complex was isolated by either anti-FLAG or anti-EGFP antibody from cell extracts. Immunoprecipitation of EGFP-Prdm12 co-isolated the FLAG-G9a complex and vice versa (Fig. 2A). Next we examined whether Prdm12 could bind to other H3K9 HKMTases (here we used GLP/Ehmt1/KMT1D and ESET/Setdb1/KMT1E), but no interactions between EGFP-Prdm12 and FLAG-GLP or FLAG-ESET were detected by Co-IP analysis (Fig. 2B). These results indicated that Prdm12 specifically forms a complex with G9a.

The second ZF domain of Prdm12 is required for G9a association and HKMTase activity. (A) Co-immunoprecipitation (Co-IP) of G9a and Prdm12. HEK293T cells were co-transfected with plasmids encoding EGFP-Prdm12, FLAG-G9a or both. Top, anti-FLAG blots for G9a. Bottom, anti-GFP blots for EGFP-Prdm12. (B) Co-IP of EGFP-Prdm12 with H3K9 methyltransferase indicated above (FLAG-G9a, FLAG-GLP, or FLAG-ESET). Cell lysates from HEK293T cells transfected with a combination of plasmids were subjected to IP using an anti-GFP antibody. Immunoprecipitates were then subjected to western blot analysis with anti-FLAG (top) or anti-GFP (bottom) antibody. (C) Schematic diagram of FLAG-Prdm12 and mutants. Numbers indicate the amino acids that were replaced. Point mutation (*), deletion (\/). (D) Co-IP determined that the ZF domains of Prdm12 interacted with G9a. HEK293T cells were co-transfected with plasmids encoding G9a and FLAG-Prdm12 or the mutants described in (C). Lysates were immunoprecipitated with anti-FLAG antibodies. Immunoprecipitates were then subjected to western blot analysis with anti-G9a (top) or anti-FLAG (bottom) antibodies. (E) FLAG-Prdm12 or mutants were transfected to HEK293T cells then immunoprecipitated with anti-FLAG antibodies. Immunoprecipitates were subjected to the in vitro methylation assay as described in Fig. 1. Methylated Histone H3 (top). The amounts of proteins were revealed by coomassie brilliant blue (bottom). ⋆, FLAG-Prdm12 and mutants.
To define which Prdm12 domain interacts with G9a, we used FLAG-Prdm12 to make PR or ZF domain deletion mutants (FLAG-Prdm12ΔPR, FLAG-Prdm12ΔZF) and a series of point mutants (Fig. 2C). In the point mutants, we replaced G115 or F117 (two conserved sites in PR domain) with Alanine (FLAG-Prdm12G115A, FLAG-Prdm12F117A) and disrupted the structure of the first or second ZF domains by replacing the two cysteines in C2H2 with arginine (FLAG-Prdm12Z1-, FLAG-Prdm12Z2-). We used these mutants and G9a in the co-immunoprecipitation assays. Except for FLAG-Prdm12ΔZF and FLAG-Prdm12Z2-, FLAG-Prdm12 and other mutants bound to G9a (Fig. 2D). These results showed that the second zinc finger domain is necessary for Prdm12 to associate with G9a.
We then determined whether the HKMTase activity of Prdm12 is correlated with the association with G9a. To this end, FLAG-Prdm12 and mutants were isolated from HEK293T cells for an in vitro methylation assay. The FLAG-Prdm12ΔZF and FLAG-Prdm12Z2- complexes lacking G9a binding capacities could not methylate histone H3 (Fig. 2E). We concluded that the Prdm12 HKMTase activity depends on the association with G9a through the second zinc finger domain.
Induction of Prdm12 by RA in P19 CellsBecause Prdm12 is expressed in the nervous system from the early embryogenesis stage (Kinameri et al., 2008) and because Prdms have functions in cell differentiation during the developmental processes (Fog et al., 2012), we sought to investigate the possible role of Prdm12 in neurogenesis. P19 embryonic carcinoma cells are a well-established in vitro model for the study of neural differentiation. In this model, P19 cells were first cultured in bacteria grade dishes for aggregation with 1 μM RA. After 4 days, the cells were transferred to a cell culture dish and the media was changed to RA-free media. After 1 day, 10% serum media were replaced with 0.5% serum media for final differentiation (Fig. 3A, top). Firstly, the mRNA expression of Prdm12 was determined by quantitative RT-PCR during this differentiation process. In response to RA, Prdm12 mRNA expression was increased at day 2 and reached a high point at day 4 then decreased following the withdrawal of RA (Fig. 3A, bottom, triangle). Aggregation of the P19 cells without RA caused a temporary increase in Prdm12 mRNA expression at day 2; however, the mRNA expression decreased on day 4 (Fig. 3A, bottom, closed circle). This indicated that RA increased and sustained Prdm12 mRNA expression. To monitor the RA-induced neural differentiation of P19, the mRNA expressions of the pluripotent transcription factor Oct3/4 and neuronal Class III β-Tubulin (TuJ1, neural-specific differentiation marker) were determined using quantitative RT-PCR (Fig. 3A, bottom). As expected, the expression of Oct3/4 mRNA was decreased, and that of Tuj1 was increased. Because Prdm12 interacts with G9a, we also determined the mRNA and protein expression of G9a during the differentiation process. Like the expression pattern of Prdm12 mRNA, the expression of G9a mRNA increased at days 2 and 4, then decreased at day 5. A point of difference, however, was that mRNA expression of G9a increased again at day 7 (Fig. 3A, bottom). While the trend of G9a protein expression was similar to that of G9a mRNA expression, GLP protein expression remained stable during differentiation (Fig. 3B).

RA induces the expression of Prdm12 in P19 cells. (A) Top, schematic diagram of the protocol for RA-induced neural differentiation in P19 cells. The details are mentioned in the Materials and Methods. Bottom, Prdm12, G9a, Oct3/4 and Tuj1 mRNA levels were determined by quantitative RT-PCR during the differentiation process. 0.1%V EtOH, negative control. Error bars represent the s.d., * p<0.05 versus EtOH control at day 4. (B) The expression of the G9a and GLP proteins was detected by western blot analysis at the indicated time point. α-tubulin was used as an internal control. (C) Immunoprecipitation-western blot to detect RA-induced Prdm12 proteins. P19 cells were cultured in bacterial grade dishes or cell culture dishes with or without 1 μM RA for 4 days. Cell lysates were subjected to immunoprecipitation followed by western blot analysis both with anti-Prdm12 antibody. IgG: Negative control for Immunoprecipitation. ⋆, Predicted size of Prdm12 (40 kDa).
To further confirm the induction of Prdm12 by RA at the protein level, P19 cells were cultured in cell culture dishes or bacteria grade dishes with or without RA for 4 days. IP-western blot analysis revealed that Prdm12 proteins were detected in response to RA regardless of aggregation or not (Fig. 3C, lane 6 and 8). Correlated with the quantitative RT-PCR results, at day 4 Prdm12 proteins were not detected in the P19 cell suspension in the absence of RA treatment (Fig. 3C, lane 7). These results suggested that RA elevates Prdm12 mRNA and protein expression in P19 cells.
Anti-proliferative effects of Prdm12 on P19 CellsBased on the RA-induced expression of Prdm12 in P19 cells, we speculated that Prdm12 may play a role in this neuronal differentiation model. To examine this hypothesis, we made two shRNAs (shPrdm12#1 and shPrdm12#2) to knock down Prdm12 expression and one scrambled shRNA as a negative control. After infection and puromycin selection, we checked the knockdown efficiency of stable Prdm12 knockdown P19 cells (P19/shPrdm12) by quantitative RT-PCR or IP-western blotting. RA-induced mRNA and protein expression were efficiently repressed in the P19/shPrdm12 cells (Fig. 4 A, and B). We then used these P19/shPrdm12 cells for RA-induced neuronal differentiation. Surprisingly, during the first aggregation stages we observed that under RA treatment P19/shPrdm12 cells formed more embryonic bodies than control cells. We directly counted the cell numbers at day 4; P19/shPrdm12 cells had about twice the number of cells than the control cells (Fig. 4C). After differentiation induced, we calculated the percentage of mature neurons by immunocytochemistry (here we used Tuj1 as a marker of mature neurons); however, there was no significant difference in the percentage of Tuj1-positive neurons in P19/shPrdm12 cells and in control P19 cells (Fig. S1).
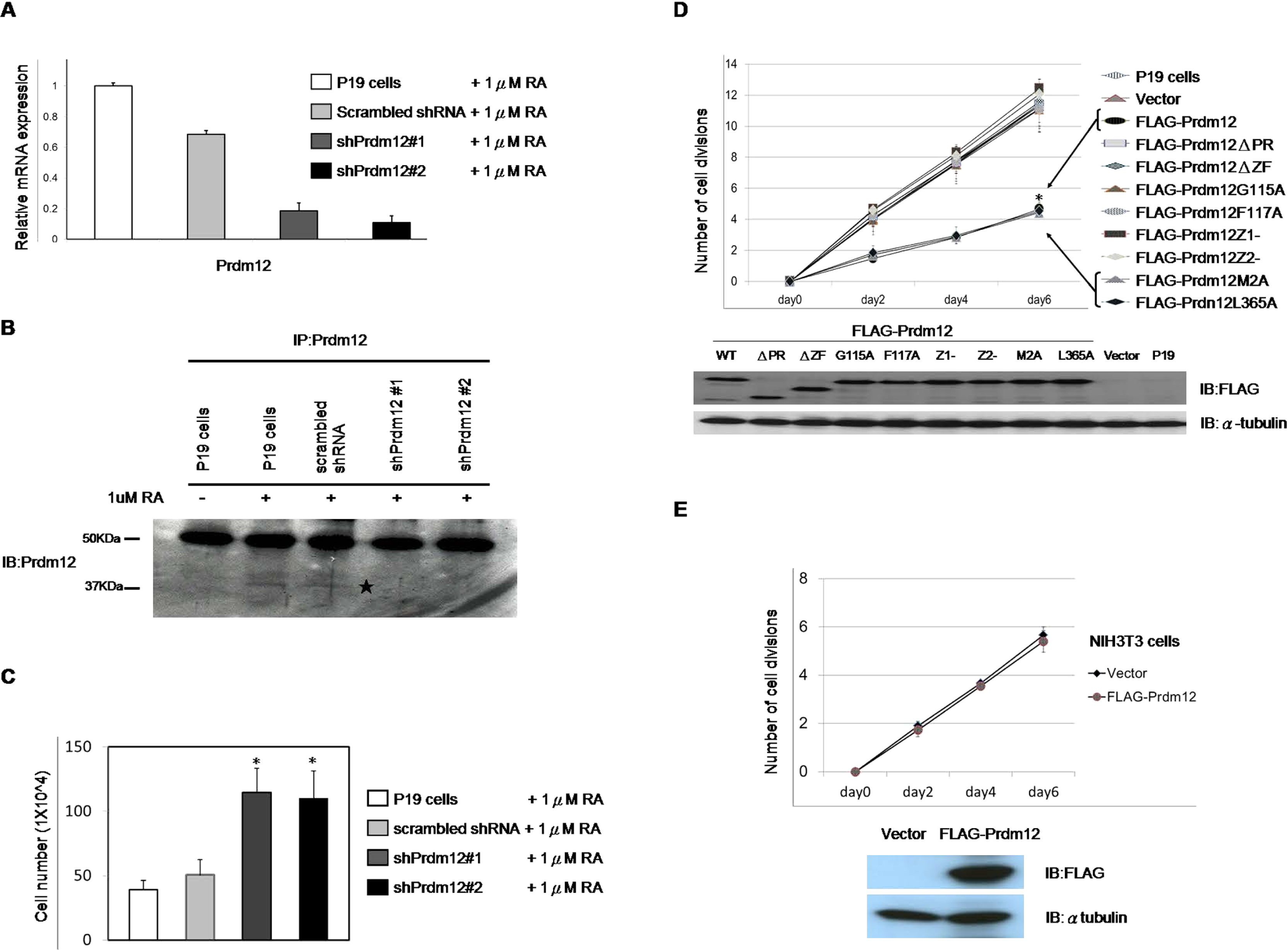
Prdm12 decreases cell proliferation in P19 cells. (A–B) Knockdown of Prdm12 in P19 cells. P19 cells were infected with indicated shRNA vectors. The stable Prdm12 knockdown cells incubated with 1 μM RA for 96 h in bacteria grade dishes. Quantitative RT-PCR (A) or western blotting (B) were performed to detect the knockdown efficiency of shPrdm12. ⋆, Predicted size of Prdm12 (40 kDa). (C) 1×106 of indicated cells were seeded in bacteria grade dishes with 1 μM RA. After 96 h, cells were trypsinized to count viable cells by Trypan Blue vital dye. Bars, SD. * p<0.05 versus scrambled shRNA control. (D) Growth curves of FLAG-Prdm12-overexpressing P19 cells. P19 cells were transfected with the indicated vectors. After selection, 1×105 pool cells were seeded onto 6 well plates and counted every 48 h for 6 days. The growth curve was presented in cell division times (top). Bars, SD. * p<0.05 versus control cells. Expression levels of FLAG-Prdm12 and mutants were detected by western blot with anti-FLAG antibodies for Prdm12 protein (bottom) and α-tubulin as the internal control. (E) Prdm12 does not affect cell proliferation in NIH-3T3 cells. NIH-3T3 cells were transfected with an empty construct or a construct expressing FLAG-Prdm12. After selection, cell growth curves of stable FLAG-Prdm12-expressing NIH-3T3 cells and control cells were determined as described in (D). Bars, SD. Expression of FLAG-Prdm12 was detected by western blot analysis with an anti-FLAG antibody.
To further examine the effect of Prdm12 on the growth of P19 cells, FLAG-Prdm12 constructs and mutants were transfected into P19 cells to produce stable overexpressing cell lines (P19/FLAG-Prdm12, P19/mutants). Besides the mutants described earlier, we made two point mutations: the amino acids at the furthest N- or C-termini were mutated to be used as controls (FLAG-Prdm12M2A, FLAG-Prdm12L365A, Fig. 2C). We analyzed cell growth by counting the number of cells for 6 days to calculate the cell division times. Compared to P19 cells, the cell division times of the P19/FLAG-Prdm12 cells decreased about 50% at day 6 (Fig. 4D). P19/FLAG-Prdm12M2A and P19/FLAG-Prdm12L365A cells still grew as slowly as the P19/FLAG-Prdm12 cells; however, the other P19/mutants cells (with the deletions or point mutations in the PR or ZF domains) lost the ability to inhibit cell proliferation. To determine whether the ability of Prdm12 to impair cell proliferation is cell type specific, we used NIH-3T3 cells instead of P19 cells. The cell growth rates were similar between the Prdm12-overexpressing NIH-3T3 cells and the control cells (Fig. 4E). Taken together, these results suggested that Prdm12 specifically impairs cell proliferation in the P19 cells through the PR and ZF domains.
To determine the location of Prdm12, we directly observed the distribution of Prdm12 and the mutants in the P19 cells by immunocytochemistry. Only FLAG-Prdm12ΔZF moved to the cytoplasm while the other mutants remained in the nucleus as FLAG-Prdm12 (Fig. 5). This data suggested that some Prdm12 mutants lose the ability to decrease cell proliferation, which may not result from mislocalization.
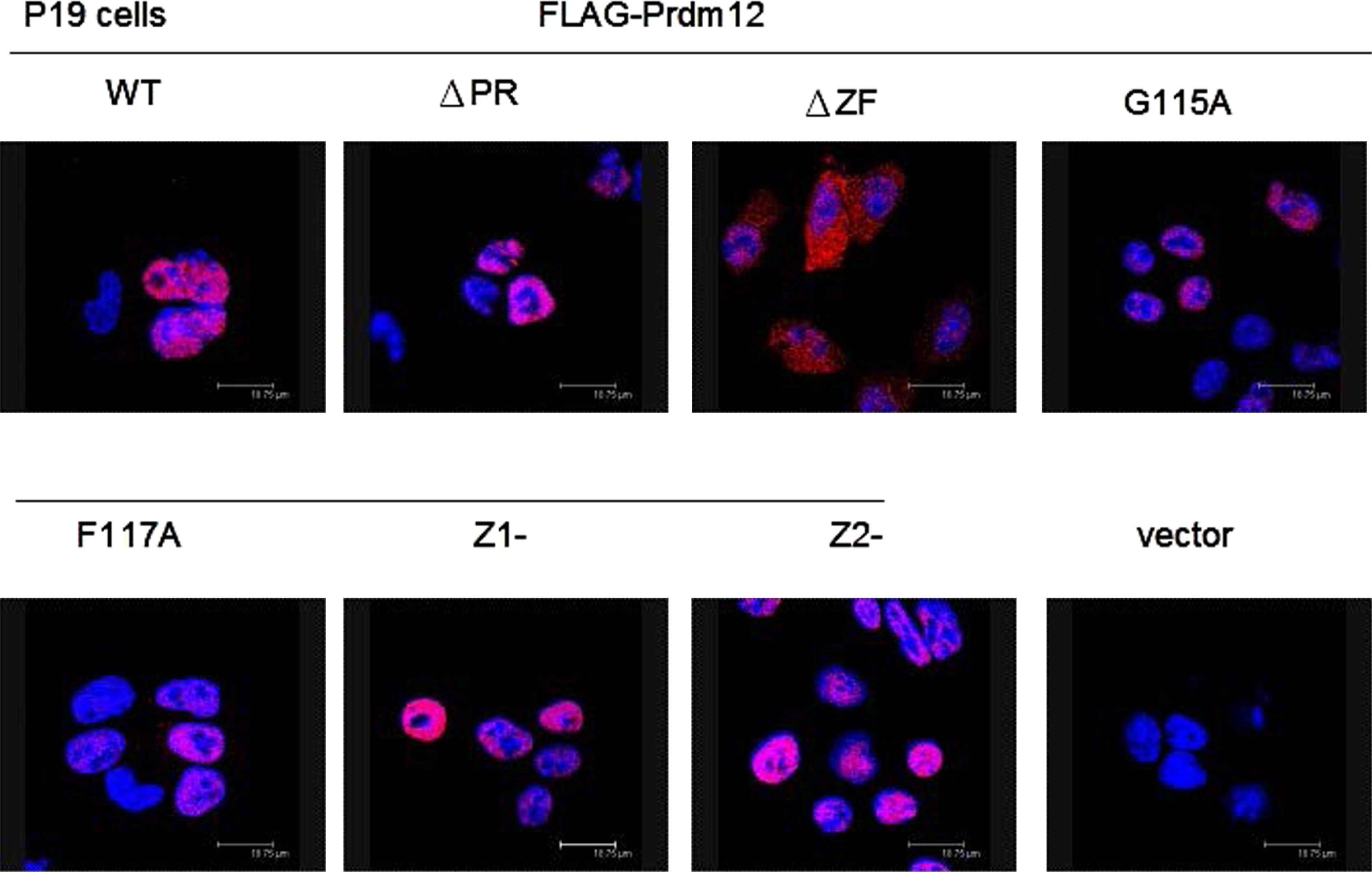
Nuclear localization of Prdm12. Plasmids expressing FLAG-Prdm12 or mutants were transfected into P19 cells. After fixation, cells were immunostained with anti-FLAG antibodies and labeled by goat anti-mouse IgG Alexa 568 secondary antibody for overexpressed proteins (red). DAPI staining (blue) for cell nucleus. Images were visualized by confocal microscopy.
Next, we investigated the pathway through which Prdm12 regulated the proliferation of P19 cells. Because cell death was not increased in the P19/Prdm12 cells (data not shown), we examined whether Prdm12 slows cell growth through control of the cell cycle. To this end, we analyzed the cell cycle distribution of P19 cells expressing FLAG-Prdm12 or deletion mutants by flow cytometry. Cell cycle analysis revealed that, compared to the control, the G1 population of P19/FLAG-Prdm12 cells was increased from 20.6% to 25.7%. FLAG-Prdm12ΔPR and FLAG-Prdm12ΔZF did not change the cell cycle distribution (Table I). This result suggested that the ability of Prdm12 to decrease the proliferation of P19 cells may occur partially through cell cycle regulation of the G1 phase.
| % G1 | % S | % G2/M | |
|---|---|---|---|
| P19 cells | 20.67 (3.66) | 57.62 (7.04) | 19.93 (3.83) |
| Vector | 20.62 (2.49) | 57.77 (4.66) | 19.55 (3.24) |
| Flag-PRDM12 | 25.77 (2.85)* | 56.08 (3.41) | 15.47 (1.70) |
| Flag-PRDM12ΔPR | 20.33 (1.81) | 58.1 (6.07) | 20.07 (5.15) |
| Flag-PRDM12ΔZF | 20.22 (2.72) | 58.9 (5.80) | 19.07 (4.15) |
1×106 cells expressing indicated proteins were plated to 10-cm cell culture dishes for 24 h. Cell cycle was measured by flow cytometry as described in Materials and Methods. *, p<0.05 versus control cells.
The G1 to S phase transition is inhibited by cyclin-dependent kinase (CDK) inhibitory proteins (CKIs). CKI families have two sub-families: the INK4 family and the CIP/KIP family. To determine whether Prdm12 increased the G1 population through upregulation of CKIs, we chose one CKI protein from each family respectively to examine expression levels in Prdm12- or mutant-overexpressing P19 cells by western blot analysis (INK4 family, p15; CIP/KIP family, p27). The protein expression of p15 was the same but the expression of p27 was increased in the P19/FLAG-Prdm12 cells (Fig. 6). As revealed by quantitative RT-PCR, the expression of p27 mRNA was also upregulated in P19/FLAG-Prdm12 cells (data not shown). The expressions of P15 and p27 were not changed in the deletion mutant-overexpressing P19 cells. These data indicated that Prdm12 upregulates p27 expression. This therefore shows that either the PR or ZF domains of Prdm12 are necessary but not sufficient for the anti-proliferation function of Prdm12.

Upregulation of p27 by overexpressing Prdm12 in P19 cells. When cells were harvested for the cell cycle analysis as described in Table I, a proportion of the cells were subjected to western blot analysis with anti-p15, anti-p27, and anti-FLAG antibodies for p15, p27, and the overexpressed Prdm12 proteins. α-tubulin was used as an internal control. The expression of p15 was not changed. p27 was increased in P19/FLAG-Prdm12 cells.
It has been reported that RA increases the number of G1 phase cells and the expression of p27 in P19 cells (Sasaki et al., 2000; Pao et al., 2011). In this paper, we described that ectopic Prdm12 has the same function in P19 cells and that RA could induce the expression of Prdm12. There are putative RARβ-response elements in the promoter region of Prdm12 predicted by transcription-element search system analysis (data not shown). Furthermore, Prdm12 is expressed in the dorsal root ganglia (DRG) of the mouse embryo (Kinameri et al., 2008) and RARβ2 is upregulated by RA in embryonic DRG neurons (Corcoran et al., 2000). Considering these findings, we hypothesized that Prdm12 may play a role downstream of RARβ2 in the RA signaling pathway. To examine this hypothesis, RARβ2 antagonists or analogues could be applied. This direction is worth further investigation, because RA induces axon outgrowth in embryonic DRG neurons by RARβ2 and the RA signaling pathway is activated in injured neurons (Wong et al., 2006). In addition, Prdm14 is required for the axon growth of primary motor neurons in zebrafish (Liu et al., 2012). If Prdm12 is transcriptionally activated by RARβ2, it may have a role in this axon regeneration pathway.
Prdm14 maintains the human ES cell identity and repressed expression of differentiation marker genes (Tsuneyoshi et al., 2008; Chia et al., 2010). Prdm12 only decreases cell proliferation in P19 cells but not NIH3T3 cells (Fig. 4). Because P19 cells are embryonic carcinoma cells, we also tested the anti-proliferative function of Pdm12 in mouse ES cells (TT2 cell lines). Overproduction of Prdm12 also results in a cell growth defect in TT2 cells (data not shown). Correlated with the expression of Prdm12 in mouse embryo, we suggest that Prdm12 regulates a pathway specifically active in “stem” cells.
Here we demonstrate with an in vitro model that Prdm12 interacts with G9a to mediate HKMTase activity (Fig. 2). Prdm1 also functions to recruit G9a through ZF domains, and G9a activity is required for Prdm1 to repress the transcription of target genes (Gyory et al., 2004). If Prdm12 regulates transcription and G9a or/and G9a-mediated H3K9 dimethylation (a silencing mark) is also involved in this process, Prdm12 targeted to the promoter region would correlate with transcriptional repression. Therefore, we hypothesize that Prdm12 may indirectly increase the expression of p27 by repressing the transcription of negative regulator(s) of p27. It has been reported recently that retinal progenitor cells lacking G9a show defects in differentiation and proliferation, but photoreceptor precursors lacking G9a still develop into normal retina (Katoh et al., 2012). This data indicates that G9a has a stage- or lineage-specific role(s) in proliferation and differentiation, and that it may be involved in the Prdm12-mediated function(s) of neuronal cells.
ZF domains are not only required for G9a interactions, Fig. 5 shows that the Prdm12 ZF domain contains nuclear localization signals (NLS) for its nuclear localization. Although the PR domain is not needed for H3K9 methylation, both the PR domain and ZF domains are necessary for Prdm12 to decrease cell proliferation in a gain-of-function model and each domain alone is not sufficient for Prdm12 (Fig. 4). It has been reported that the PR and ZF domains are required for Prdm4 to inhibit the cell cycle through the transcriptional repression of cyclin E (Chittka et al., 2004). Therefore, the properties of Prdm12 we demonstrated are similar to other Prdms.
Although Prdm12 is widely expressed in the nervous system, its function is still unknown. We hope our new findings will provide some insights into the role(s) of Prdm12 in neural development.
We thank current and former Shinkai lab members, especially Mikiko Fukuda, Tae Komai, and Makoto Tachibana for their technical support and scientific discussions. This study was supported in part by a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.