Abstract
The NACK kinesins (NACK1, NACK2 in tobacco and AtNACK1/HINKEL, AtNACK2/STUD/TETRASPORE in Arabidopsis), members of a plant-specific kinesin-7 family, are required for cytokinesis. Previous studies using tobacco and Arabidopsis cells showed that NACK1 and AtNACK1 at the phragmoplast midzone activate the MAP kinase cascade during the late M phase, which is critical for the cell plate formation. However, the loss-of-function phenotype has not been investigated in details in living cells and the molecular activity of this kinesin remains to be determined. Here, we report the mitotic roles and activity of the NACK kinesins in the moss Physcomitrella patens. When we simultaneously knocked down three PpNACKs by RNA-interference (RNAi) in protonemal cells, we observed a cytokinesis failure following a defect in phragmoplast expansion. In addition, misaligned chromosomes were frequently detected in the pre-anaphase spindle and the anaphase onset was significantly delayed, indicating that PpNACK also plays a role in pre-anaphase. Consistent with the appearance of early and late mitotic phenotypes, endogenous PpNACK was localised to the interpolar microtubule (MT) overlap from prometaphase through telophase. In vitro MT gliding assay and single motor motility assay showed that PpNACK-b is a processive, plus-end-directed motor, suggesting that PpNACK is capable of transporting cargoes along the spindle/phragmoplast MT. Our study using Physcomitrella patens demonstrated that PpNACK is an active motor protein and identified unexpected and conserved roles of PpNACK during the mitosis of P. patens.
Introduction
Kinesin superfamily proteins play a variety of roles in spindle formation, chromosome segregation, and cytokinesis (Cross and McAinsh, 2014; Goshima and Vale, 2003). Since the majority of these mitotic kinesins generate force towards spindle microtubules (MTs) and chromosomes, identification of their loss-of-function phenotypes and precise molecular activities have led to the construction of a model that describes the mechanism of these processes. The biochemical and cell biological characterisations have been extensively, or in some instances nearly completely, performed in several model animal and yeast cell types (Goshima and Vale, 2003, 2005; Hildebrandt and Hoyt, 2000; Maliga et al., 2013; Tytell and Sorger, 2006). On the other hand, the complete picture has not been drawn for plant kinesins, partly because plants encode many more kinesins in their genome, many of which are associated with the mitotic apparatus (Miki et al., 2014).
The kinesin-7 family is a large family in plants, which is further divided into four subfamilies (kinesin 7-I to kinesin 7-IV). In the moss Physcomitrella patens, seven genes belong to this family (Shen et al., 2012). Kinesin 7-III is most homologous to a well-studied animal kinetochore kinesin, CENP-E, and similarly to CENP-E, P. patens kinesin 7-III (one member) is localised to the kinetochore from prophase to anaphase (Miki et al., 2014). Kinesin 7-IV (one member) is localised to the spindle and phragmoplast MT; the function of this motor is not known (Miki et al., 2014). On the other hand, mitotic localisation has not been observed for kinesin 7-I (two members).
P. patens kinesin 7-IIs are orthologous to the NACK kinesins (NACK1, NACK2 in tobacco and AtNACK1/HINKEL, AtNACK2/STUD/TETRASPORE in Arabidopsis) (Nishihama et al., 2002; Strompen et al., 2002; Yang et al., 2003). Previous studies in tobacco cells have shown that NACK1 is localised to the phragmoplast equator, recruits and activates MAPKKK (NPK1) and thereby the MAPK cascade. This pathway, named the NACK-PQR pathway, is required for cytokinesis (Nishihama et al., 2002; Sasabe and Machida, 2012; Soyano et al., 2003). Specifically, the non-motor region of NACK1 was shown to interact with NPK1, supporting a model that NACK kinesin transports NPK1 along the phragmoplast MTs towards the equator (Ishikawa et al., 2002; Nishihama et al., 2002). However, the motor activity of the NACK kinesin subfamily has not been determined.
Another intriguing observation in flowering plants is that NACK1 kinesin and MAPKKK are the direct effectors of cell cycle regulation. Cyclin-dependent kinases (CDKs) phosphorylate NACK1 and MAPKKK and inhibit their interaction during early M phase, suggesting a mechanism by which activation of the MAPK cascade is inhibited until the cytokinesis phase (Sasabe et al., 2011). A substrate of MAPK is the conserved MT bundling protein MAP65; phosphorylation of MAP65 during late M phase attenuates its MT bundling activity, which leads to phragmoplast MT disassembly that is required for cell plate expansion (Sasabe et al., 2006).
In P. patens, three highly homologous genes belong to the kinesin 7-II subfamily (hereafter labelled PpNACK-a, PpNACK-b, and PpNACK-c) (Fig. S1). PpNACKs are expressed and localised to the phragmoplast equator in the tissue called ‘protonemata’, which has two types of cells, ‘chloronemata’ and ‘caulonemata’ (Miki et al., 2014). This localisation is consistent with tobacco and Arabidopsis orthologues. However, live imaging of the endogenous PpNACKs tagged with Citrine (a green fluorescent protein [GFP] variant) indicated that they are also localised to the midzone of the metaphase spindle, suggesting that PpNACKs have additional roles in the metaphase (Miki et al., 2014).
In this study, we utilised protonemal cells of P. patens to elucidate the mitotic role of PpNACK. P. patens is an emerging model land plant for cell and developmental biology (Cove, 2005; Cove et al., 2006; Hiwatashi et al., 2008; Nakaoka et al., 2012; Prigge and Bezanilla, 2010; Vidali et al., 2009). The rate of homologous recombination is very high in P. patens, such that gene disruption and GFP tagging of the endogenous gene are easy to perform. A conditional RNAi system was also recently developed, in which a knockdown of a single or multiple homologous genes can be induced in protonemal cells (Kosetsu et al., 2013; Miki et al., 2014, 2015; Nakaoka et al., 2012). Furthermore, live cell microscopy can be easily performed in protonemal cells, which are found in a single layer and actively grow and divide. Using P. patens protonemal cells, we showed that PpNACKs play critical roles in chromosome alignment in the metaphase plate. Furthermore, in the absence of PpNACKs, MT interdigitation of the phragmoplast MTs was abnormal, and phragmoplast MTs disappeared without expansion. Finally, the PpNACK motor showed processive, plus-end-directed motility along MTs.
Materials and Methods
Plasmids, moss culture, RNAi, and transformation
For moss culture and transformation, we followed the previously described methods (Miki et al., 2014; Nakaoka et al., 2012). In brief, regular culturing of protonemata was carried out on a BCDAT medium with cellophane (Futamura Chemical Industries Co., Ltd, Nagoya, Japan) at 25°C under continuous white light. Transformation was performed by the standard polyethylene glycol (PEG)-mediated method using protoplast; driselase (Kyowa Hakko Bio Co., Ltd., Tokyo, Japan) was used for cell wall digestion. To observe RNA-interference (RNAi) phenotypes, we cultured protonemata for 3–4 days in the presence of 1 μM β-estradiol, which turns on the promoter and thereby the expression of dsRNAs. Protonemal cells, which had been cultured on a glass-bottom (MatTek, Ashland, MA, USA) or 6-well glass-bottom (IWAKI, Japan) plates with BCD agar medium at 24–25°C, were imaged. Tobacco NACK1 was subcloned into the vector that contains the constitutive EF1α promoter.
The method to establish stable RNAi lines for PpNACKs is described in Miki et al. (2015). Both GFP-tubulin and histoneH2B-mRFP were expressed in these lines. RNAi constructs targeting the ORF region were made by using the Gateway system (Invitrogen, Carlsbad, CA, USA) with pGG626 (Nakaoka et al., 2012) as the destination vector (PCR primers used for RNAi are listed in Table I). In some cases, the pGG624 plasmid in which the RFP-RNAi cassette was deleted from pGG626 was used as the alternative destination vector. Transient RNAi was performed using previously described method (Kosetsu et al., 2013). Briefly, the RNAi constructs were transformed into the GFP-tubulin/histone-mRFP line or the wild-type using the PEG-mediated method. Transformed cells were selected by hygromycin (30 mg/L). RNAi was induced by 1 μM β-estradiol. After ~4 d, the cells were fixed with 8% paraformaldehyde and stained with 0.005% calcofluor and 10 μg/mL propidium iodide. pKK133 (MAP65 RNAi) was used as a positive control.
Table I
PCR primer sequences used to construct transgenic lines and expression plasmids
| Plasmid name |
Forward primer (5'→3') |
Reverse primer (5'→3') |
Accession number (Phypa #) |
Locus name |
| 1. RNAi constructs |
| 1st (PpNACK-a) |
CACCgcggccgcATTCGCAACTAGGGCGAAGG |
ATAggcgcgccGTGGCGCGAAGGCTGTGCATTG |
458197 |
Pp1s343_28V6 |
| 2nd (PpNACK-a) |
CACCgcggccgcCAAATCCGACCTTAATTCAAGC |
ATAggcgcgccGATCCTCGCTACCACTTCAGC |
458197 |
Pp1s343_28V6 |
| 3rd (PpNACK-b) |
CACCgcggccgcGTAACACCCTTGCATTTGC |
ATAggcgcgccGCTGTGGCACGAAGGTTGTGC |
432536 |
Pp1s43_22V6 |
| 4th (PpNACK-c) |
CACCgcggccgcTAATCAAAATTTGCGAAGC |
ATAggcgcgccACCTCTTCTTCCTCCAGC |
454208 |
Pp1s255_53V6 |
| 5th (PpNACK-a) |
TATAgcggccgcAAAAAAagatctGTTTCCCTGCGGTTCCCGTAGCTC |
TATAggcgcgccAAAAAAggatccAGGCCTACTACGTAGGTTCTCCTT |
458197 |
Pp1s343_28V6 |
| 6th (PpNACK-b) |
TATAgcggccgcAAAAAAagatctGCTTTGTTTCCTTCCCTGCCTTTG |
TATAggcgcgccAAAAAAggatccGGCTGACAATAGAGGCCTCTTATG |
432536 |
Pp1s43_22V6 |
| 7th (PpNACK-c) |
TATAgcggccgcAAAAAAagatctGACAGATCGAGGGAGCGAGCGAG |
TATAggcgcgccAAAAAAggatccGGCTGTGGACGCTAAGCCACCC |
454208 |
Pp1s255_53V6 |
| 2. Motor constructs for in vitro assays |
| PpNACK-b, fragment-1* |
AAAcatatgACTGTGGCCATTGAGAGAGG |
GATTTGATGCGATCTAGAGGACAC |
432536 |
Pp1s43_22V6 |
| PpNACK-b, fragment-2* |
GTGTCCTCTAGATCGCATCAAATC |
AAAAgcggccgcAGTTCTCTTGGCTGCCTCTTCTGC |
432536 |
Pp1s43_22V6 |
| 3. Mis12-mCherry integration plasmid |
| PpMis12-C terminus |
ATAgggcccGAGCAAGTTCATCGAAGAAGATC |
ATCaagcttGAGTGCATCGCTTGTATCAGCAGG |
70890 |
Pp1s30_96V6 |
| PpMis12-3'UTR |
AAAggatccTAAGATCGAAGCTCTCTAAAGAGAAG |
TTTactagtTATATTTGCAAAAGTATACTGATG |
70890 |
Pp1s30_96V6 |
Primers constructed for the 5th and 6th constructs were used for the 8th construct, and those for the 5th–7th for the 9th construct. 1st–4th constructs were made in Miki et al. (2015).
*Fragments 1 and 2 were ligated with the XbaI site (underlined). The restriction enzyme sites were indicated with lower-case letters.
Long-term fluorescence imaging and fixed cell observation of the RNAi lines, was carried out with Ti microscope (10× 0.45-NA, 10× 0.50-NA, or 20× 0.75-NA lens) (Nikon, Tokyo, Japan) equipped with an electron-multiplying charge-coupled device (EMCCD) camera (Evolve, Photometrics, Tucson, AZ, USA; or iXon3, Andor Technology Ltd., Belfast, UK). For live imaging, images were acquired at multiple sites every 3 min for >10 h. High-magnification of the time-lapse microscopy of the PpNACK-b-Citrine or RNAi lines, was achieved with a Nikon TE2000 microscope equipped with a spinning-disc confocal unit CSU-X1 (Yokogawa Electric Corporation, Tokyo, Japan) and an EMCCD camera with a ×100 1.40-NA lens (ImagEM; Hamamatsu Photonics, K.K., Hamamatsu, Japan). Images were acquired every 30 s. For in vitro total internal reflection fluorescence microscopy (TIRFM) assay, Nikon’s Ti microscope (×100 1.49-NA lens) equipped with an EMCCD camera (Evolve) and 488/561-nm excitation laser was used. The microscopes were controlled by the Micromanager software (Edelstein et al., 2010) or NIS elements (Nikon), and image data were analysed with ImageJ.
Protein purification
Motor and dimerisation domains of PpNACK-b were fused with mGFP and 6×His, and cloned into an E. coli expression vector. Primers listed in Table I were used for PCR, and cloning was performed with NdeI/XbaI/NotI digestion. We basically followed the procedure described in Miki et al. (2015). Proteins were expressed in E. coli SoluBL21 strain (Genlantis, San Diego, CA, USA) at 18°C with 0.2 mM IPTG. Bacterial pellet from 250 mL culture was homogenised in lysis buffer (50 mM NaPO4, pH 8.0, 250 mM NaCl, 2 mM MgCl2, 20 mM imidazole, 10 mM 2-mercaptoethanol, 1 mM ATP, protease inhibitors), mixed with Ni-NTA beads, washed with wash buffer (50 mM NaPO4, pH 6.0, 250 mM NaCl, 1 mM MgCl2, 10 mM 2-mercaptoethanol, 0.1 mM ATP), followed by elution (50 mM NaPO4, pH 7.2, 250 mM NaCl, 1 mM MgCl2, 500 mM imidazole, 10 mM 2-mercaptoethanol, 0.1 mM ATP). The MT gliding and kinesin motility assays were performed immediately after protein purification, because the activity could not be maintained on ice or in the freezer.
MT gliding assay and kinesin motility assay
The conventional MT gliding assay (Woehlke et al., 1997) was performed with some modifications (Miki et al., 2015). Briefly, flow chambers were assembled using a coverslip and a microslide glass with double-sided sticky tape. The coverslip was coated with the anti-GFP antibody. The chamber was washed with BRB80 and the purified mGFP-tagged PpNACK-b motor was loaded into the chamber. After washing with BRB80 containing 0.5 mg/mL k-casein, the chamber was loaded with motility buffer prepared from BRB80, GMPCPP-stabilised MTs with Alexa 568-labels, 2 mM ATP or AMP-PNP, 0.5 mg/mL k-casein, and 0.1% methylcellulose, 20 μM taxol with oxygen scavenger system (50 mM glucose, 400 μg/mL glucose-oxidase, 200 μg/mL catalase, and 4 mM DTT) (Li et al., 2012). To prepare polarity-marked and GMPCPP-stabilised MTs, bright MT seeds (80% unlabelled tubulin: 20% Alexa 568-labelled tubulin) were first assembled, followed by polymerisation of dim MTs (67% unlabelled tubulin: 2% Alexa 568-labeled tubulin: 31% NEM [N-ethylmaleimide]-tubulin). For the kinesin motility assay (Tomishige and Vale, 2000), a silanized coverslip was coated with the anti-biotin antibody. Solution containing 1% pluronic acid was then loaded into the chamber. After one wash with BRB80, GMPCPP-stabilised MTs labelled with Alexa 568 and biotin were loaded in the presence of taxol. After a single wash with BRB80, PpNACK-b-mGFP was added to the motility buffer.
Results
RNAi of PpNACKs causes a metaphase delay as well as cytokinesis failure
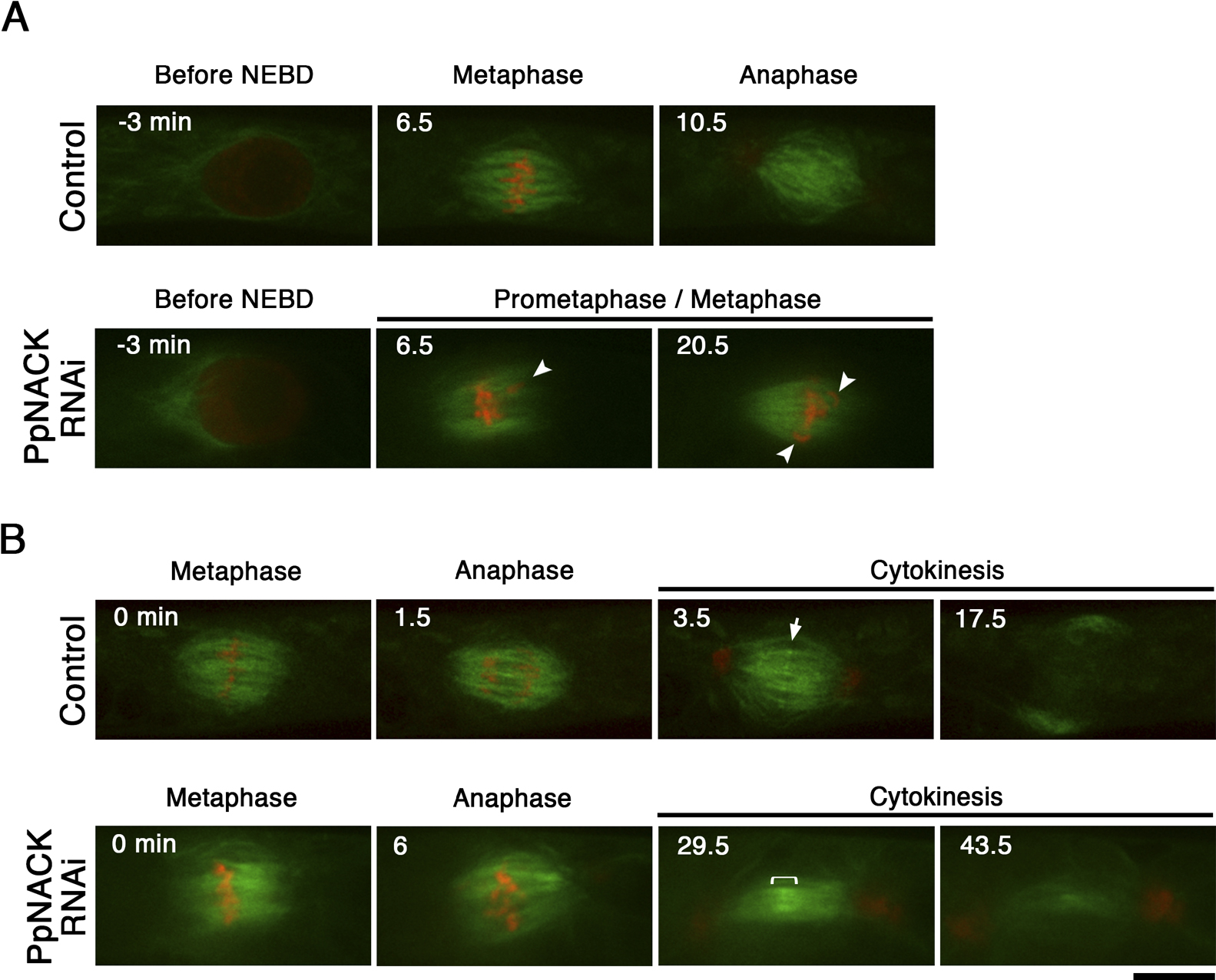
To systematically survey the role of each kinesin in moss, we recently performed an RNAi screening targeting >60 types of P. patens kinesins (Miki et al., 2015). The screening identified characteristic mitotic defects—cytokinesis failure and mitotic delay—for multiple RNAi constructs targeting PpNACKs (Fig. 1A–D, Movie 1). Since PpNACKs are highly homologous to each other, it was expected that one construct targeted multiple PpNACK paralogues. The appearance of identical phenotype for a total of five constructs, including three non-overlapping RNAi constructs strongly suggested that the phenotype was derived from a knockdown of PpNACKs rather than as an off-target effect of the RNAi construct.

To test redundancy of the three PpNACK genes, we performed ‘transient RNAi’ by transforming the RNAi vector targeting the 5'UTR sequences, which are not well conserved among the PpNACK genes (Fig. 1B, Fig. 2A). This assay was suitable for observation of nuclear numbers, which are indicator of the cytokinesis failure (Kosetsu et al., 2013) (however, we did not succeed in time-lapse mitosis imaging). Transformation of a single RNAi vector targeting PpNACK-a or PpNACK-b alone did not give rise to the binuclear phenotype (<10%). However, double knockdown of PpNACK-a and -b resulted in higher frequency of binucleated cells (24.2±6.8%, n=3 replicates; a total of 97 observed colonies), and the transformation of the a–c triple knockdown construct maximised the defect (36.0±9.0%, n=4 replicates; a total of 103 observed colonies) (Fig. 2A). These results suggest that PpNACK-a, -b, and -c function redundantly in protonemal cells, which is similar to the function of Arabidopsis’ NACKs (HINKEL and TETRASPORES) (Oh et al., 2008; Tanaka et al., 2004).

When the transient RNAi treatment of three PpNACKs was combined with ectopic expression of tobacco NACK1, the binuclear cells were significantly reduced (Fig. 2B). This cross-species rescue result further supported the notion that the PpNACK RNAi phenotype was derived from a knockdown of PpNACKs.
Chromosome alignment is impaired in the absence of PpNACKs
We observed the dynamics of the pre-anaphase spindle (GFP-tubulin) and chromosomes (histoneH2B-mRFP) in the absence of PpNACKs using spinning-disc confocal microscopy (Fig. 3). We used two independent triple-RNAi transgenic lines and identified identical phenotypes. In all cells that showed a prometaphase delay, we observed chromosome misalignment in the bipolar spindle, suggesting that the delay is caused by the mitotic checkpoint that monitors proper attachment of MTs to the kinetochore and/or tension generated at the kinetochore (n=11; Fig. 3A, Movie 2). We concluded that PpNACK plays a role in chromosome alignment, which is a novel phenotype for this kinesin subfamily.
Phragmoplast expansion is inhibited in the absence of PpNACKs
As was expected from the longer time-lapse imaging (Fig. 1C), many cells that had misaligned chromosomes eventually entered anaphase and formed the phragmoplast. However, in ~50% of cells, lagging chromosomes appeared in the anaphase spindle, and the phragmoplast morphology and dynamics were abnormal (Fig. 3B). In control cells, interdigitation of the phragmoplast MTs was clearly observed at the equator, as indicated by the sharply elevated GFP-tubulin signals (Fig. 3B, arrow; Movie 3) (Hiwatashi et al., 2008; Nakaoka et al., 2012). However, in the RNAi-treated cells, the overlap was broader. In control cells, the phragmoplast gradually expanded towards the cell wall by destabilising the interior MTs and generating new MTs at the edge. In contrast, phragmoplast expansion was completely suppressed in the RNAi-treated cells, and the phragmoplast MTs were destabilised (Fig. 3B; Movie 3). We concluded that PpNACK is required for proper interdigitation of the phragmoplast MTs as well as phragmoplast expansion.
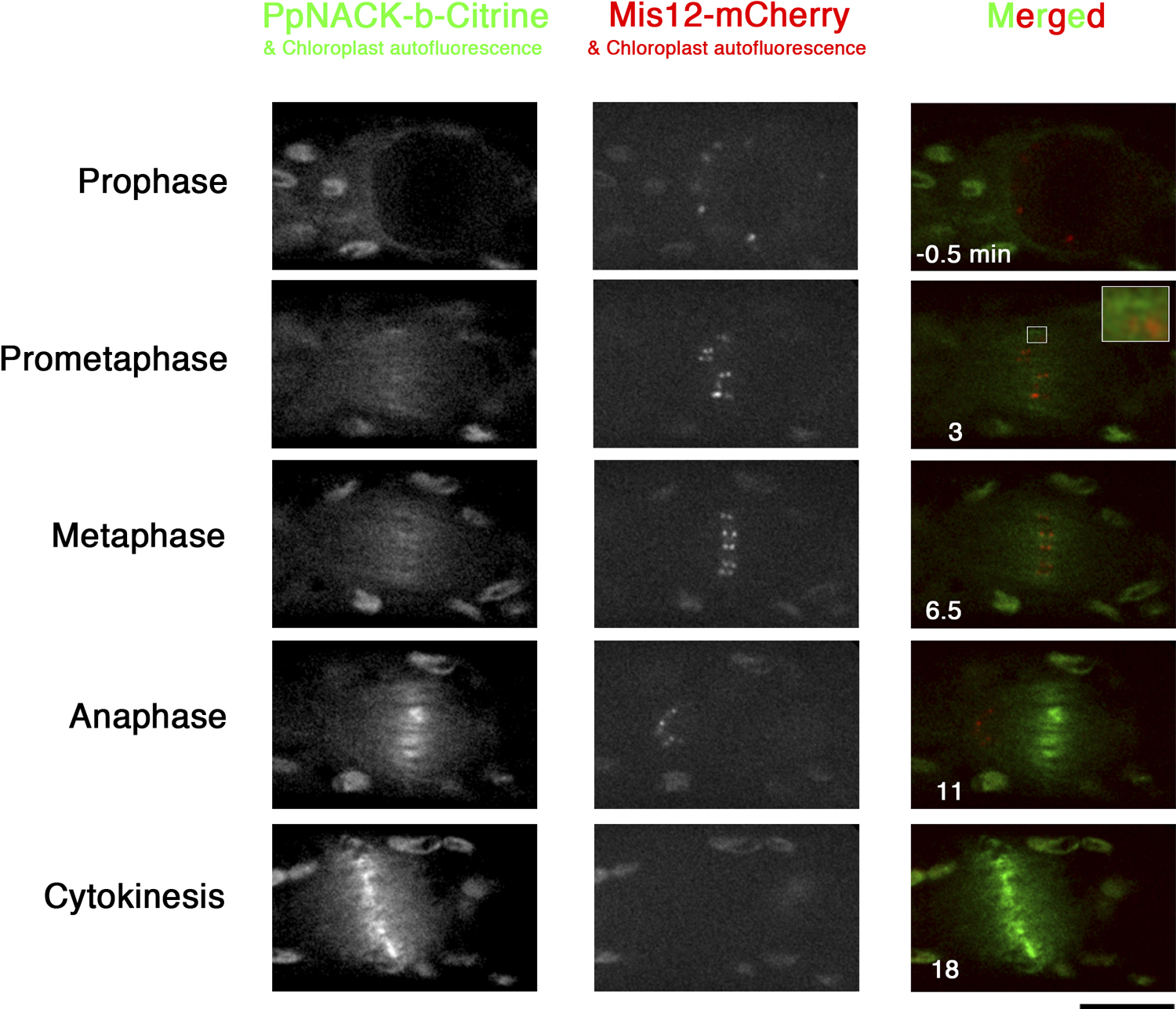
PpNACKs associate with midzone MTs from prometaphase to telophase
In tobacco cells, NACK1 is distributed throughout the cytoplasm until the anaphase. However, in our previous systematic localisation study, we described that PpNACK-a to -c were localised to the midzone from metaphase (Miki et al., 2014). To precisely determine the timing of the midzone localisation, we constructed the PpNACK-b-Citrine/PpMis12-mCherry line (Mis12 is a conserved kinetochore protein (Goshima et al., 2003)), and filmed mitosis using spinning-disc confocal microscope (Fig. 4; Fig. S2; Movie 4). We found that the discrete Citrine signals, which probably corresponded to the midzone antiparallel MT overlaps, are observed right after the NEBD (i.e. early prometaphase), at which kinetochores are yet to be aligned (Fig. 4, inset). Those signals persisted during metaphase and increased upon anaphase onset, as previously described (Miki et al., 2014). This result is consistent with the phenotypic data that PpNACK is required in both pre- and post-anaphase.
The motor activity of the NACK kinesin has not been investigated in previous studies. To test if PpNACK has the plus-end-directed motor activity, we performed a MT gliding assay using purified truncated PpNACK-b that possesses the motor and dimerisation domains (454 a.a.) (Fig. 5A, B). In this assay, the kinesin motor tagged with mGFP was bound to the glass surface via the anti-GFP antibody, and fluorescence-labelled MTs near the glass surface were observed with the TIRFM. The polarity of the MTs was marked based upon the differential labelling of the plus and minus ends (Fig. 5C, E). We observed MT gliding towards the minus-ends in the presence of ATP at the rate of 460±97 nm/s (SD; n=150; Fig. 5D, E; Movie 5). In contrast, MT gliding was never observed when ATP was replaced with AMP-PNP, a non-hydrolysable analogue of ATP (Movie 6). We concluded that PpNACK-b is a MT plus-end-directed motor protein.

Next, we performed a kinesin motility assay by observing a single PpNACK-b-mGFP motor on the MT (Fig. 5F). We found that PpNACK-b-mGFP moves along the MT at the mean velocity of 416±91 nm/s (SD; n=29; Fig. 5G, H; Movie 7). These results indicate that PpNACK-b is a processive, plus-end-directed motor protein.
Discussion
This study using P. patens protonemal cells confirmed the function of NACK in cytokinesis. Although phragmoplast dynamics were not investigated in previous studies using tobacco and Arabidopsis cells, the observed defect in phragmoplast expansion in this study is consistent with the observations in tobacco and Arabidopsis cells in which the cell plate is not expanded in the absence of NACKs. We further found the lack of sharp overlaps of MTs at the equator. These phenotypes might be explained by the mis-regulation of MAP65, a critical factor to interdigitate antiparallel MTs at the phragmoplast equator (Kosetsu et al., 2013), the activity of which was shown to be regulated by MAPK phosphorylation (Sasabe et al., 2006). The biochemical activity identified in this study also supports the idea that PpNACK transports MAPKKK along phragmoplast MTs towards the equator, where the downstream MAPK phosphorylates MAP65 and other midzone factors. However, it is also possible that PpNACK functions as a force generator at the overlapping zone independent of activation of the MAPK signalling. It remains to be clarified to what extent the PpNACK phenotype is attributed to the mislocalisation of MAPKKK and altered phosphorylation status of MAP65.
The appearance of the pre-anaphase phenotype after PpNACK RNAi was unexpected. Previous studies in tobacco showed no specific localisation of NACK1 before anaphase, and furthermore, identified CDK phosphorylation of NACK1, which inhibited interaction with MAPKKK (NPK1) (Nishihama et al., 2002; Sasabe et al., 2011). If a similar regulation involving CDK is present for PpNACK, our results imply the MAPKKK-independent function of PpNACK in ensuring integrity of the pre-anaphase spindle. Alternatively, regulation by CDK might have evolved only in flowering plants. In such a case, the NACK-PQR pathway in P. patens may be active throughout mitosis at the midzone and in phosphorylation of factors important for chromosome alignment. The RNAi transgenic lines established in this study would serve as a good system to test these hypotheses. It is noteworthy that the misalignment phenotype was caused by a factor enriched at the spindle midzone (i.e. MT overlapping region). In animal cells, chromosome misalignment is normally caused by defective kinetochore-MT interaction or misregulation of the kinetochore MT plus-end dynamics, which is controlled by kinetochore-enriched proteins (e.g. (Goshima et al., 2007)). However, PpNACKs did not accumulate at the kinetochore during prometaphase and metaphase. Together with our previous finding that none of the known MT depolymerising kinesins were accumulated at the kinetochore (Miki et al., 2014), an intriguing possibility arises that non-kinetochore MTs also make a significant contribution to chromosome alignment in P. patens.
In summary, this study identified two distinct functions of NACK during mitosis, chromosome alignment and phragmoplast expansion. Furthermore, the motor activity of this kinesin subfamily was demonstrated.
Acknowledgments
We thank Tomohiro Miki, Ken Kosetsu, Takashi Moriwaki, Ami Ito, Yuki Nakaoka, Moé Yamada, Yuji Hiwatashi, Masaki Ishikawa, Mitsuyasu Hasebe, Erik Jonsson, Michiko Sasabe, and Michio Tomishige for technical advice and valuable discussion as well as for providing reagents; Tomoko Nishiyama for her help in microscopy; Rie Inaba and Momoko Nishina for technical assistance. This work was supported by the grant from the Human Frontier Science Program and the Toray Foundation (to G.G.).
Author contributions
H.N. and G.G. designed the research; H.N. performed experiments; H.N. and G.G. analysed the data; H.N. and G.G. wrote the paper.
References
- Cove, D. 2005. The moss Physcomitrella patens. Annu. Rev. Genet., 39: 339–358.
- Cove, D., Bezanilla, M., Harries, P., and Quatrano, R. 2006. Mosses as model systems for the study of metabolism and development. Annu. Rev. Plant Biol., 57: 497–520.
- Cross, R.A. and McAinsh, A. 2014. Prime movers: the mechanochemistry of mitotic kinesins. Nat. Rev. Mol. Cell Biol., 15: 257–271.
- Edelstein, A., Amodaj, N., Hoover, K., Vale, R., and Stuurman, N. 2010. Computer control of microscopes using microManager. Curr. Protoc. Mol. Biol., Chapter 14: Unit14 20.
- Goshima, G., Kiyomitsu, T., Yoda, K., and Yanagida, M. 2003. Human centromere chromatin protein hMis12, essential for equal segregation, is independent of CENP-A loading pathway. J. Cell Biol., 160: 25–39.
- Goshima, G. and Vale, R.D. 2003. The roles of microtubule-based motor proteins in mitosis: comprehensive RNAi analysis in the Drosophila S2 cell line. J. Cell Biol., 162: 1003–1016.
- Goshima, G. and Vale, R.D. 2005. Cell cycle-dependent dynamics and regulation of mitotic kinesins in Drosophila S2 cells. Mol. Biol. Cell, 16: 3896–3907.
- Goshima, G., Wollman, R., Goodwin, N., Zhang, J.M., Scholey, J.M., Vale, R.D., and Stuurman, N. 2007. Genes required for mitotic spindle assembly in Drosophila S2 cells. Science, 316: 417–421.
- Hildebrandt, E.R. and Hoyt, M.A. 2000. Mitotic motors in Saccharomyces cerevisiae. Biochim. Biophys. Acta, 1496: 99–116.
- Hiwatashi, Y., Obara, M., Sato, Y., Fujita, T., Murata, T., and Hasebe, M.. 2008. Kinesins are indispensable for interdigitation of phragmoplast microtubules in the moss Physcomitrella patens. Plant Cell, 20: 3094–3106.
- Ishikawa, M., Soyano, T., Nishihama, R., and Machida, Y. 2002. The NPK1 mitogen-activated protein kinase kinase kinase contains a functional nuclear localization signal at the binding site for the NACK1 kinesin-like protein. Plant J., 32: 789–798.
- Jones, D.T., Taylor, W.R., and Thornton, J.M. 1992. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci., 8: 275–282.
- Katoh, K., Misawa, K., Kuma, K., and Miyata, T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res., 30: 3059–3066.
- Katoh, K. and Standley, D.M. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol., 30: 772–780.
- Kosetsu, K., de Keijzer, J., Janson, M.E., and Goshima, G. 2013. MICROTUBULE-ASSOCIATED PROTEIN65 is essential for maintenance of phragmoplast bipolarity and formation of the cell plate in Physcomitrella patens. Plant Cell, 25: 4479–4492.
- Li, W., Moriwaki, T., Tani, T., Watanabe, T., Kaibuchi, K., and Goshima, G. 2012. Reconstitution of dynamic microtubules with Drosophila XMAP215, EB1, and Sentin. J. Cell Biol., 199: 849–862.
- Maliga, Z., Junqueira, M., Toyoda, Y., Ettinger, A., Mora-Bermudez, F., Klemm, R.W., Vasilj, A., Guhr, E., Ibarlucea-Benitez, I., Poser, I., Bonifacio, E., Huttner, W.B., Shevchenko, A., and Hyman, A.A. 2013. A genomic toolkit to investigate kinesin and myosin motor function in cells. Nat. Cell Biol., 15: 325–334.
- Miki, T., Naito, H., Nishina, M., and Goshima, G. 2014. Endogenous localizome identifies 43 mitotic kinesins in a plant cell. Proc. Natl. Acad. Sci. USA, 111: E1053–1061.
- Miki, T., Nishina, M., and Goshima, G. 2015. RNAi screening identifies the armadillo repeat-containing kinesins responsible for microtubule-dependent nuclear positioning in Physcomitrella patens. Plant Cell Physiol., DOI: 10.1093/pcp/pcv002.
- Nakaoka, Y., Miki, T., Fujioka, R., Uehara, R., Tomioka, A., Obuse, C., Kubo, M., Hiwatashi, Y., and Goshima, G. 2012. An inducible RNA interference system in Physcomitrella patens reveals a dominant role of augmin in phragmoplast microtubule generation. Plant Cell, 24: 1478–1493.
- Nishihama, R., Soyano, T., Ishikawa, M., Araki, S., Tanaka, H., Asada, T., Irie, K., Ito, M., Terada, M., Banno, H., Yamazaki, Y., and Machida, Y. 2002. Expansion of the cell plate in plant cytokinesis requires a kinesin-like protein/MAPKKK complex. Cell, 109: 87–99.
- Oh, S.A., Bourdon, V., Das ’Pal, M., Dickinson, H., and Twell, D. 2008. Arabidopsis kinesins HINKEL and TETRASPORE act redundantly to control cell plate expansion during cytokinesis in the male gametophyte. Mol. Plant, 1: 794–799.
- Prigge, M.J. and Bezanilla, M. 2010. Evolutionary crossroads in developmental biology: Physcomitrella patens. Development, 137: 3535–3543.
- Sasabe, M., Soyano, T., Takahashi, Y., Sonobe, S., Igarashi, H., Itoh, T.J., Hidaka, M., and Machida, Y. 2006. Phosphorylation of NtMAP65-1 by a MAP kinase down-regulates its activity of microtubule bundling and stimulates progression of cytokinesis of tobacco cells. Genes Dev., 20: 1004–1014.
- Sasabe, M., Boudolf, V., De Veylder, L., Inze, D., Genschik, P., and Machida, Y. 2011. Phosphorylation of a mitotic kinesin-like protein and a MAPKKK by cyclin-dependent kinases (CDKs) is involved in the transition to cytokinesis in plants. Proc. Natl. Acad. Sci. USA, 108: 17844–17849.
- Sasabe, M. and Machida, Y. 2012. Regulation of organization and function of microtubules by the mitogen-activated protein kinase cascade during plant cytokinesis. Cytoskeleton (Hoboken), 69: 913–918.
- Shen, Z., Collatos, A.R., Bibeau, J.P., Furt, F., and Vidali, L. 2012. Phylogenetic analysis of the Kinesin superfamily from physcomitrella. Front. Plant Sci., 3: 230.
- Soyano, T., Nishihama, R., Morikiyo, K., Ishikawa, M., and Machida, Y. 2003. NQK1/NtMEK1 is a MAPKK that acts in the NPK1 MAPKKK-mediated MAPK cascade and is required for plant cytokinesis. Genes Dev., 17: 1055–1067.
- Strompen, G., El Kasmi, F., Richter, S., Lukowitz, W., Assaad, F.F., Jurgens, G., and Mayer, U. 2002. The Arabidopsis HINKEL gene encodes a kinesin-related protein involved in cytokinesis and is expressed in a cell cycle-dependent manner. Curr. Biol., 12: 153–158.
- Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol., 28: 2731–2739.
- Tanaka, H., Ishikawa, M., Kitamura, S., Takahashi, Y., Soyano, T., Machida, C., and Machida, Y. 2004. The AtNACK1/HINKEL and STUD/TETRASPORE/AtNACK2 genes, which encode functionally redundant kinesins, are essential for cytokinesis in Arabidopsis. Genes Cells, 9: 1199–1211.
- Tomishige, M. and Vale, R.D. 2000. Controlling kinesin by reversible disulfide cross-linking. Identifying the motility-producing conformational change. J. Cell Biol., 151: 1081–1092.
- Tytell, J.D. and Sorger, P.K. 2006. Analysis of kinesin motor function at budding yeast kinetochores. J. Cell Biol., 172: 861–874.
- Vidali, L., Rounds, C.M., Hepler, P.K., and Bezanilla, M. 2009. Lifeact-mEGFP reveals a dynamic apical F-actin network in tip growing plant cells. PLoS One, 4: e5744.
- Woehlke, G., Ruby, A.K., Hart, C.L., Ly, B., Hom-Booher, N., and Vale, R.D. 1997. Microtubule interaction site of the kinesin motor. Cell, 90: 207–216.
- Yang, C.Y., Spielman, M., Coles, J.P., Li, Y., Ghelani, S., Bourdon, V., Brown, R.C., Lemmon, B.E., Scott, R.J., and Dickinson, H.G. 2003. TETRASPORE encodes a kinesin required for male meiotic cytokinesis in Arabidopsis. Plant J., 34: 229–240.