SPIRAL2 Stabilises Endoplasmic Microtubule Minus Ends in the Moss Physcomitrella patens
2018 年 43 巻 1 号 p. 53-60

詳細
2018 年 43 巻 1 号 p. 53-60
Stabilisation of minus ends of microtubules (MTs) is critical for organising MT networks in land plant cells, in which all MTs are nucleated independent of centrosomes. Recently, Arabidopsis SPIRAL2 (SPR2) protein was shown to localise to plus and minus ends of cortical MTs, and increase stability of both ends. Here, we report molecular and functional characterisation of SPR2 of the basal land plant, the moss Physcomitrella patens. In protonemal cells of P. patens, where non-cortical, endoplasmic MT network is organised, we observed SPR2 at minus ends, but not plus ends, of endoplasmic MTs and likely also of phragmoplast MTs. Minus end decoration was reconstituted in vitro using purified SPR2, suggesting that moss SPR2 is a minus end-specific binding protein (-TIP). We generated a loss-of-function mutant of SPR2, in which frameshift-causing deletions/insertions were introduced into all four paralogous SPR2 genes by means of CRISPR/Cas9. Protonemal cells of the mutant showed instability of endoplasmic MT minus ends. These results indicate that moss SPR2 is a MT minus end stabilising factor.
Key words: acentrosomal microtubule network, microtubule minus end, P. patens, CAMSAP/Nezha/Patronin
Microtubules (MTs) are dynamic polymers consisting of α- and β-tubulins. In vivo, their plus ends show characteristic dynamics with five distinct processes: polymerisation, depolymerisation, catastrophe, rescue, and pausing (Akhmanova and Steinmetz, 2008; Desai and Mitchison, 1997). In contrast, minus ends are often stabilised and static (Akhmanova and Hoogenraad, 2015; Toya and Takeichi, 2016). γ-Tubulin, the major cellular MT nucleator, protects minus ends by capping (Wiese and Zheng, 2000). In animals, CAMSAP/Nezha/Patronin has been identified as another minus end stabilising/protecting protein, which recognises minus ends and inhibits their growth and shortening directly (Goodwin and Vale, 2010; Jiang et al., 2014). CAMSAPs play roles in various cellular processes such as metaphase spindle morphogenesis (Goshima et al., 2007), anaphase spindle elongation (Wang et al., 2013), cell adhesion (Meng et al., 2008), cell polarisation (Toya et al., 2016), or cell migration (Jiang et al., 2014), illustrating the significance of minus end stabilisation/protection in a variety of intracellular processes where MTs are involved (Akhmanova and Hoogenraad, 2015; Toya and Takeichi, 2016).
MT minus ends have to be stabilised also in plants that do not possess centrosomes. However, genes homologous to CAMSAPs are not present in the plant genome. Recently, the plant-specific protein SPIRAL2 (SPR2), mutants of which are defective in directional cell elongation and exhibit right-handed helical growth in longitudinally expanding organs (Buschmann et al., 2004; Furutani et al., 2000; Shoji et al., 2004), was identified as the MT minus end stabiliser in plants (Fan et al., 2018; Nakamura et al., 2018). In its absence, minus ends of cortical MTs are depolymerised more rapidly, which suppresses MT severing at the crossover site, resulting in MT array disorientation (Fan et al., 2018; Nakamura et al., 2018).
Here we studied SPR2 in protonemal cells in the moss Physcomitrella patens. Protonemal cells develop endoplasmic (non-cortical) MT networks, in which individual MT ends are traceable with high-resolution microscopy.
Molecular cloning and transgenic moss selection Transgenic moss lines and PCR primers used in this study are listed in Table I and Table II, respectively. The transformation method is described thoroughly in Yamada et al. (2016). Homologous recombination was confirmed by PCR as described in Fig. 1B, C. To generate CRISPR-mediated frameshift mutations (Collonnier et al., 2017), 20 bp guide RNAs (gRNAs) targeting the start of SPR2a, SPR2b, SPR2c, SPR2d genes were designed using CRISPOR (http://crispor.tefor.net/). Forward and reverse oligonucleotides bearing the 20 bp gRNA template sequences with sticky ends were ligated into the BsaI site in pCasGuide/pUC18. Regions from PpU6 promoter to the end of gRNA scaffold were amplified by PCR and ligated into PCR-amplified pSY034 by InFusion (TAKARA Clontech) to create a gRNA multicassette plasmid. Equal amounts of circular gRNA multicassette plasmid and Streptococcus pyogenes Cas9 expression vector were simultaneously transformed into the moss. Treated protoplasts were subjected to nourseothricin selection up to 10 d after transformation, after which surviving protoplast-turned-colonies were cultivated on drug-free medium. Moss colonies were subjected to PCR amplification of SPR2a genes and subsequent DNA sequencing to identify CRISPR-mediated indels. Indels in singlets or doublets that resulted in frameshift mutations were selected for and subjected to PCR amplification and sequencing for remaining SPR2 genes to identify moss colonies that had non-triplet indels in all four SPR2 genes.
| Line # | Clone # | Name | Plasmid | Resistance | References |
|---|---|---|---|---|---|
| 0367 | 52 | mCherry-tubulin | pTM426 | BSD | Kosetsu et al., 2013 |
| 0379 | 2 | EB1-Citrine/mCherry-tubulin | pKK037 | G418 Zeo | Kosetsu et al., 2013 |
| 0595 | 2, 20 | SPR2a-Citrine/mCherry-tubulin | pMY284 | BSD G418 | |
| 0625 | 7, 21-2 | spr2 mutant/EB1-Citrine/mCherry-tubulin | pGenius-SpCas9 pSY069 | G418 Zeo |
Zeo: Zeocin, BSD: Blasticidin
| Plasmid | Description | Gene | 5’ primer/Forward primer | 3’ primer/Reverse primer | Targeting site |
|---|---|---|---|---|---|
| pMY284 | C-ter Citrine | SPR2a | AAActcgagCTCAACTTCAAGTCTGGTAAAGC AAAaagcttAAGCTTACTCTCCCCAGTATCGCCACTTC |
AAAtctagaTAGTTGGAAGTCTGGTCATCGAG AAAccgcggGTCAAAGTTTGTGAGACTTCC |
Endogenous |
| Genotyping PCR | SPR2a | CTTCCGTTTTGAAATCTGGGTGATAGG | GGTTGTTACAGGATGTCCTATTTCTTCC | ||
| Citrine | TCGCCGGACACGCTGAACTT | TCGCCGGACACGCTGAACTT | |||
| pGenius SpCas9 | Streptococcus pyogenes Cas9 expression plasmid (Collonnier et al., 2017). | ||||
| pSY034 | pCasGuide/pUC18 (Collonnier et al., 2017) was modified to replace the BsaI site with MfeI and ClaI sites. The resulting plasmid was further modified to include a nourseothricin resistance expression cassette. | ||||
| pSY069 | gRNA multicassette | SPR2a, b, c, d | cgttatgctccattaGTCTCTGCAAGACCCTTCCTCTA gtaaaacgacggccagtgccaag gtgtcaaagtcactAAGCTTCGATTGAATGTCCATTGA acactcaagagttgtAGCTTCGATTGAATGTCCATTGA gtctctgcaagacccAGCTTCGATTGAATGTCCATTGA |
taatggagcataacgGTACCCGGGGATCCTCTAGAAAA tggcactggccgtcgttttacaa tagtgactttgacacgtttaTCTAGAAAAAAAGCACCGACTCG acaactcttgagtgttaaTATCTAGAAAAAAAGCACCGACTCG gggtcttgcagagacTCTAGAAAAAAAGCACCGACTCG |
Endogenous 5’ region |
Lowercase letters indicate the restriction enzyme site or overlapping region for InFusion cloning (TAKARA Clontech).
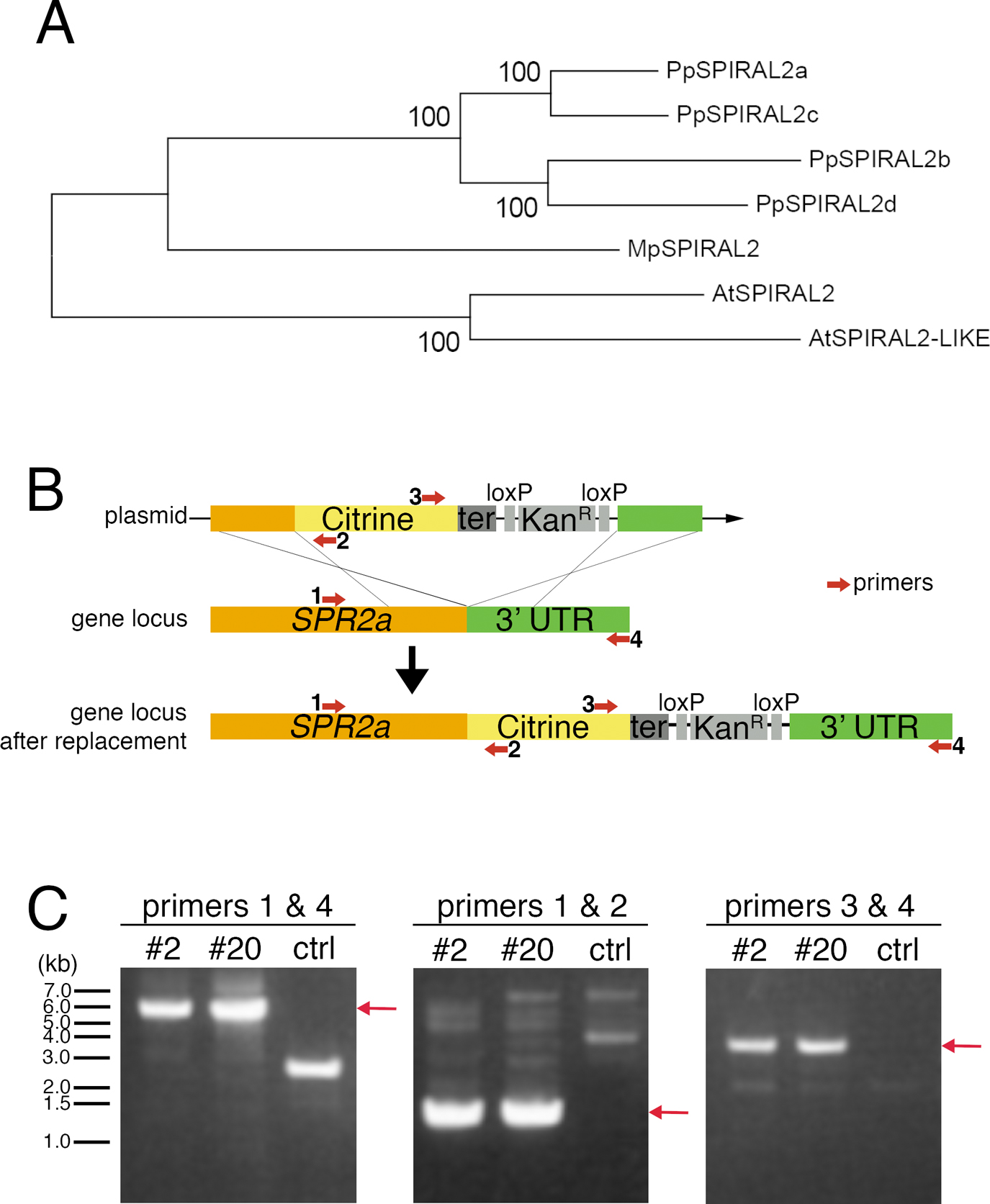
Citrine-tagging to endogenous SPR2a. (A) SPR2 phylogeny across Physcomitrella patens, Arabidopsis thaliana, and the liverwort Marchantia polymorpha. After SPR2 protein sequences were aligned with MAFFT, regions with gaps were removed from the alignment using MacClade. The phylognetic tree was constructed using neighbour joining (NJ) methods using the Molecular Evolutionary Genetics Analysis (MEGA) software. Reliability was assessed using 1000 trials in the bootstrap method. P. patens genes used in this study are found in Phytozome (http://www.phytozome.net/physcomitrella.php) as follows: SPIRAL2a (Phpat.020G028300), SPIRAL2b (Phpat.024G021900), SPIRAL2c (Phpat.023G069000), and SPIRAL2d (Phpat.008G049800). Gene IDs for other genes are as follows: A. thaliana (At)-SPIRAL2 (AT4G27060), (At)-SPIRAL2-LIKE (AT1G50890), and M. polymorpha (Mp)-SPIRAL2 (Mapoly0121s0024). (B) Schematic outline of C-terminal Citrine-tagging of SPR2a by homologous recombination. Numbered red arrows indicate PCR primers. (C) Drug selection-surviving transformed moss lines #2 and #20 were screened with colony PCR using the primers indicated in (B). Red arrows indicate expected PCR product band sizes of successfully transformed moss.
In vivo microscopy Spinning-disc confocal microscopy using 100× 1.45-NA lens and ImagEM camera (Hamamatsu Photonics) was performed as previously described (Yamada et al., 2016). Oblique illumination fluorescence microscopy was carried out with a Nikon Ti microscope with TIRF unit, a 100× 1.49-NA lens, GEMINI split view (Hamamatsu Photonics), and EMCCD camera Evolve (Roper). Sonicated moss in BCD liquid medium were cultured in a polydimethylsiloxane (PDMS) (Dow Corning TORAY) microfluidic chamber that was created by a standard soft lithography technique. The PDMS mold was fabricated by spin-coating a negative photoresist (SU-8 3025, Microchem Corp) to a thickness of 15 μm on a silicon wafer, followed by photolithography pattering of the desired microchannel layout using maskless photolithography system (DL-1000, Nano System Solutions, Inc). After the prepared PDMS layer and glass bottom dish surfaces were both treated with air plasma, they were pressed against each other and heated at 65°C for 90 min to facilitate irreversible bonding. All imaging was performed at ~23°C under dark conditions. Microscopes were controlled by Micromanager.
Data analysis Kymographs were obtained from imaging data in lines expressing mCherry-tubulin and EB1-Citrine. EB1-Citrine decorated growing MT plus ends specifically, which eliminated the possibility of mis-identifying non-growing MTs being transported in the cytoplasm as treadmilling MTs. The slope of minus ends (EB1-Citrine undecorated) was taken as shrink rate, where for kymographs with variable slopes, the least steep slope was measured. To compare how often minus end shrinkage events occurred, 5 min of 10 μm×10 μm area from 6 random individual movies taken for spr2 mutants and control lines were analysed. The count over 5 min and 100 μm2 was taken as the frequency of minus end shrinkage events; however, the frequency was underestimated, as MTs were undetectable in edges of the selected area where illumination is weaker.
Protein purification GST-SPR2a-mGFP-6xHis expression was induced in BL21 E. coli with 0.1 mM IPTG for 24 h at 18°C. Harvested cells were lysed using the Advanced Digital Sonifier D450 (Branson) in lysis buffer (50 mM Tris-HCl [pH 8.0], 100 mM KCl, 2 mM MgCl2, and 20 mM imidazole) supplemented with 10 mM β-mercaptoethanol and protease inhibitors (1 mM PMSF and peptide inhibitor cocktails). After rotation with nickel-coated beads (0.5 mL bed volume) for 2 h at 4°C, the proteins were eluted using 500 μL elution buffer (lysis buffer supplemented with 350 mM imidazole and 10 mM β-mercaptoethanol). Elution was repeated 5 to 7 times. Three most green elutes were then rotated with glutathione Sepharose beads for 2 h at 4°C, and GST was cleaved out using 8% HRV 3C protease (Thermo Fisher Scientific). Protein was flash frozen.
In Vitro MT dynamics assay We largely followed methods previously described (Li et al., 2012; Moriwaki and Goshima, 2016), which referred to (Bieling et al., 2007; Gell et al., 2010). MT growth was initiated by flowing 27 μM porcine brain tubulin (containing 8% Alexa Fluor 647-labelled tubulin) and 10 nM purified SPR2a-mGFP in assay buffer (1×MRB80, 100 mM KCl, 1 mM GTP, 0.8 μg/μL κ-casein, 0.1% methylcellulose) supplemented with an oxygen scavenger system.
P. patens possesses four highly homologous genes encoding SPR2 (SPR2a to d) (Fig. 1A). We tagged Citrine to endogenous SPR2a and observed the localisation in protonemal apical cells with confocal microscopy (Fig. 1B, C; Citrine is a YFP variant). We observed many punctate Citrine signals in the endoplasm in interphase (Fig. 2A). During mitosis, signals were similarly observed outside the nucleus before nuclear envelope breakdown (NEBD), whereas they were hardly detectable in metaphase (Fig. 2B). Punctate signals reappeared after anaphase, with many of them observed near the spindle/phragmoplast edge, where MT minus ends are highly enriched (Fig. 2B, arrowheads, Movie 1; Smertenko et al., 2017). To precisely compare the localisation of SPR2a-Citrine with MT ends, we next used oblique illumination fluorescence microscopy, with which individual cortex-proximal endoplasmic MTs are clearly visualized with minimal chloroplast autofluorescence (Nakaoka et al., 2015). We observed punctate Citrine signals at MT ends, which were slowly shrinking, while the other undecorated ends were growing (Fig. 2C, D, Movie 2). Thus, SPR2a-Citrine was localised to minus ends of endoplasmic MTs.
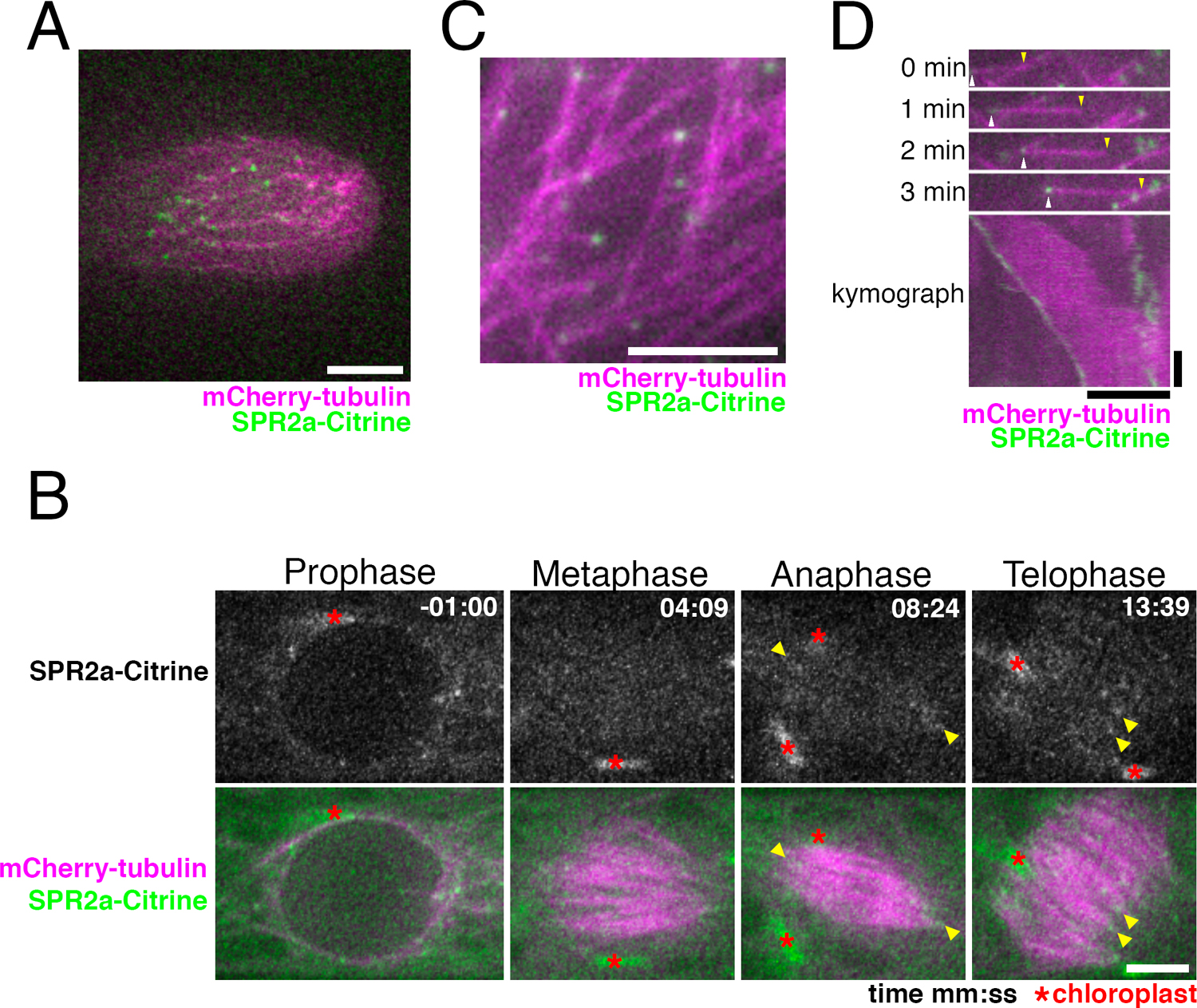
SPR2a localises to MT minus ends in vivo. (A) Punctate SPR2a-Citrine signals observed in apical regions of caulonema tip cell. Bar, 5 μm. (B) SPR2a-Citrine localisation during mitosis (time 0 corresponds to the timing of NEBD). Punctate signals are observed on MTs around the nucleus before NEBD. Signals were hardly detected at the metaphase spindle, but reappeared as the cell entered anaphase, and continued into cytokinesis (yellow arrowheads). See also Movie 1. Bar, 5 μm. (C, D) SPR2a-Citrine is observed to localise to MT minus ends with oblique illumination fluorescence microscopy. Still images and kymograph following a single MT are presented. The growing end was identified as the plus end (yellow arrowheads), while the shrinking end corresponded to the minus end (white arrowheads). Horizontal bar, 5 μm; vertical bar, 1 min.
In contrast, plus end accumulation or lattice association was not observed, unlike Arabidopsis SPR2 (Fan et al., 2018; Nakamura et al., 2018; Yao et al., 2008). AtSPR2 also accumulates at the MT crossover and regulates its stability (Fan et al., 2018; Nakamura et al., 2018; Wightman et al., 2013). However, since it is difficult to identify MT crossovers in moss protonemal cells, which have 3D MT networks, we could not assess if moss SPR2 has similar accumulation.
We concluded that moss SPR2 is specifically localised to the minus ends of MTs in the majority of the cell cycle in protonemal cells.
Purified SPR2a binds to MT minus ends in vitroPurified AtSPR2 binds to the lattice and the minus end of MTs (Buschmann et al., 2004; Fan et al., 2018; Shoji et al., 2004; Yao et al., 2008). To test if moss SPR2 has an identical biochemical activity, we aimed to purify full-length SPR2a tagged with monomeric GFP. We obtained full-length SPR2a-mGFP with two-step purification (Fig. 3A, arrow). Although other proteins and/or partially degraded SPR2a were also detected as lower molecular weight bands, our attempt of further purification with gel filtration chromatography failed: it dramatically reduced the yield of SPR2a-mGFP and also could not get rid of the other bands. We therefore used the partially purified protein obtained after two-step purification in our in vitro MT polymerisation assay.
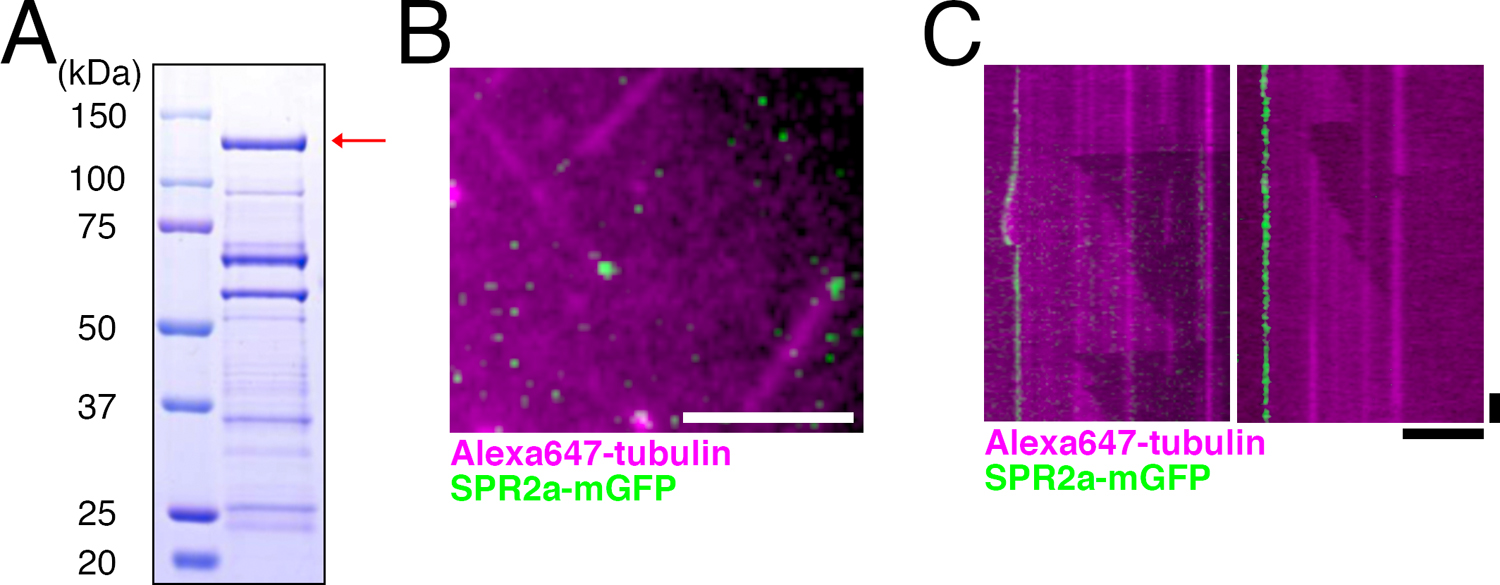
SPR2a localises to MT minus ends in vitro. (A) Coomassie blue-stained SDS-PAGE image of purified SPR2a-mGFP. Red arrow indicates expected band size of SPR2a-mGFP. Multiple other bands, some of which might represent the degradation product, were also observed. (B, C) SPR2a-mGFP (10 nM) localises to both dynamic (left) and non-dynamic (right) MT minus ends in the in vitro MT polymerisation assay. Horizontal bar, 5 μm; vertical bar, 1 min.
We first prepared stabilised MT seeds by polymerising porcine brain tubulin in the presence of non-hydrolysable GMPCPP. Tubulin dimers were then supplied together with SPR2a-mGFP protein, and their dynamics were imaged with TIRF microscopy (Fig. 3B, C). MTs were polymerised predominantly from one end of the seeds, which corresponds to the plus end. Interestingly, SPR2a-mGFP was always detected at the other ends of MTs, namely the minus ends, and their association to the lattice or plus end could not be observed (Fig. 3C). Whether SPR2a affects MT growth from the minus end could not be determined, since we rarely observed minus end MT growth in our assay, regardless of the presence or absence of SPR2a. However, we encountered several cases where SPR2a-mGFP tracked the dynamic MT minus ends, suggesting that SPR2a does not completely inhibit MT growth from the minus end (Fig. 3C, left). These data strongly suggest that moss SPR2a specifically and autonomously recognises the minus end of MTs.
Generation of the spr2 mutantWe generated a loss-of-function mutant of SPR2 by means of CRISPR/Cas9-mediated gene editing (Lopez-Obando et al., 2016) in a line stably expressing mCherry-tubulin and MT plus end-tracking EB1-Citrine (Fig. 4A). We isolated two lines, in which small indels caused frameshifts to all four SPR2 genes (lines #7 and #21-2) (Fig. 4B). Both lines developed protonemata (filamentous cells that extend from germinated spores by serial division of their apical cells), gametophores (leafy shoot-like structures that house gametangia differentiated off protonemata), and rhizoids (root-like filamentous cells differentiated from the base of gametophores) (Cove, 2005; Kofuji and Hasebe, 2014; Menand et al., 2007). Despite SPR2 being expressed in all moss organs (Ortiz-Ramirez et al., 2016), the protonema and rhizoid of spr2 mutants were not observably different from control. However, we observed undulation in the gametophore edge of spr2 mutants, suggesting that SPR2 plays a role in gametophore development (Fig. 4C).

Construction of spr2 mutants using CRISPR/Cas9-based gene editing. (A) Schematic outline of CRISPR-mediated mutagenesis of SPR2a, b, c, d genes. Imperfect repair of CRISPR-induced DSBs produces indels, where non-triple basepair indels result in frameshift mutations. (B) Genomic SPR2 sequences before and after CRISPR-mediated mutation of two individual mutant lines used in this study, #7 and #21-2. Blue line, gRNA target sequence. (C) Gametophore and rhizoid of control and spr2 mutants were not dramatically different, except for a consistent undulation in the gametophore edge of spr2 mutants (inset). Dotted line, inset outline. Bars, 1 mm (left) and 0.5 mm (right).
We observed MT dynamics in the protonemal cell of spr2 mutants using oblique illumination fluorescence microscopy (Fig. 5, Movie 3). Overall endoplasmic MT network did not look different between control and mutant cells. However, we observed an increased number of treadmilling MTs in the spr2 mutant lines, where minus ends underwent rapid shrinkage while plus ends were growing (Fig. 5A). Quantification indicated that both frequency and rate of MT minus end shrinkage were significantly increased in the spr2 mutant (Fig. 5B, C). In contrast, we could not observe a reproducible change in MT plus end growth rate. The catastrophe frequency, which is decreased in the AtSPR2 mutant (Fan et al., 2018; Nakamura et al., 2018), could not be assessed in our system, as most MTs grow out of focal plane without catastrophe. Nevertheless, we reason that the effect on plus ends, if any, is indirect in moss protonemata (e.g. due to a change in free tubulin concentration), since SPR2a was enriched exclusively at the minus end.
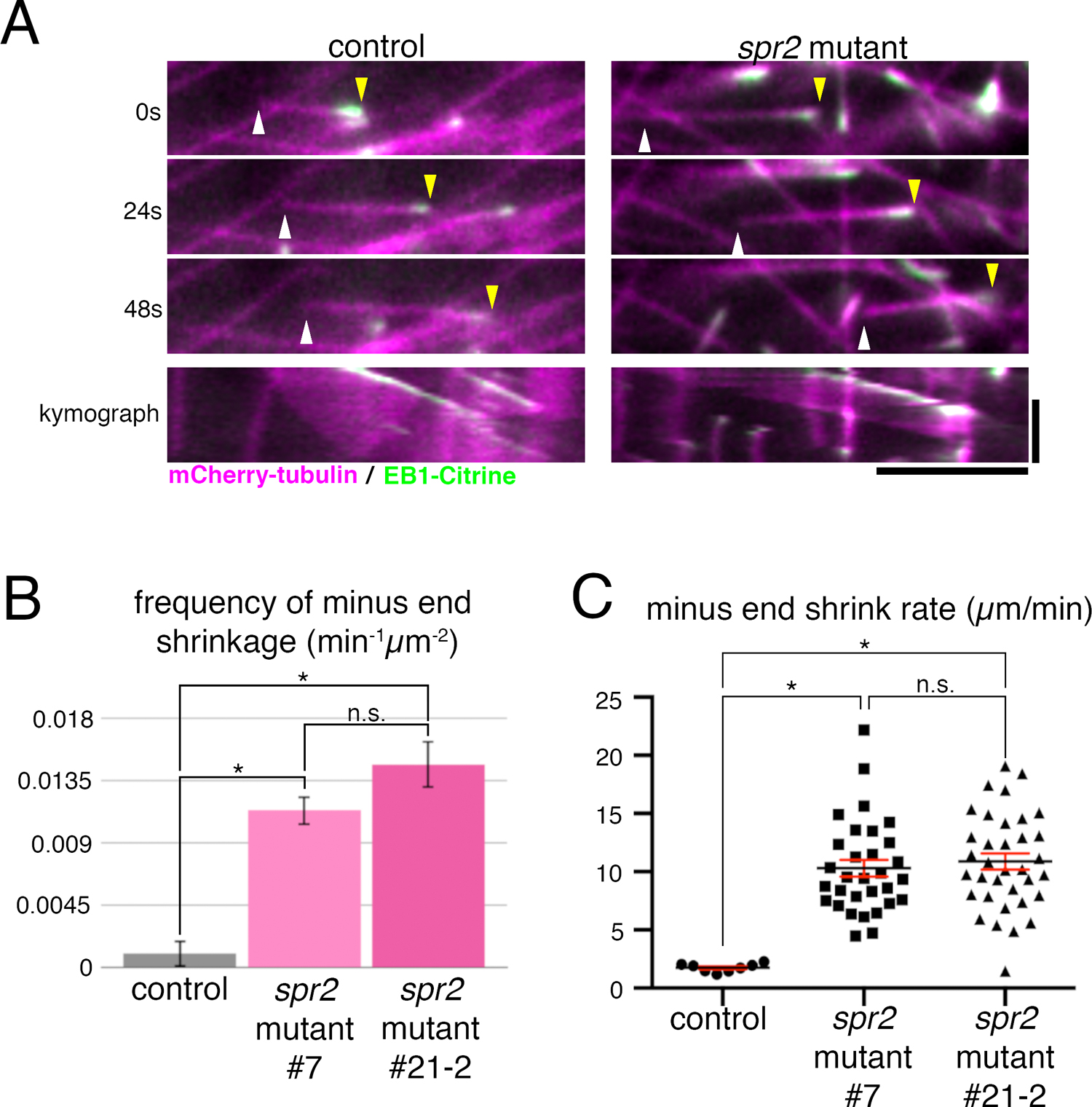
MT minus end stability is reduced in the spr2 mutant. (A) Still images and kymographs (bottom panels) taken using oblique illumination fluorescence microscopy of a treadmilling MT of control and spr2 mutant (#7) lines. The MT minus end (white arrowhead) in the spr2 mutant line shrinks faster than in the control line. Yellow arrowheads indicate MT plus ends. Horizontal bar, 5 μm; vertical bar, 1 min. (B) Frequency of minus end shrinkage in spr2 mutant lines #7 (0.0113±0.0062 min–1μm–2, mean±SD; N=6) and #21-2 (0.0147±0.0112 min–1μm–2; N=6) was significantly higher than that of the control line (0.0010±0.0017 min–1μm–2; N=6). Error bars represent SEM. Unpaired Student’s t-test assuming parametric Gaussian distribution using Welch’s correction (unassuming equal SD) was used on the data analyses. Samples were taken as significantly different when P-value was calculated to be less than 0.01. “n.s.” stands for “not significant”. (C) Minus end shrinkage rate was significantly higher in spr2 mutant lines #7 (10.30±0.72 μm/min, mean±SEM; N=31), and #21-2 (10.88±0.70 μm/min; N=35) compared to the control (1.75±0.13 μm/min; N=8). Mean (black lines) and SEM (red error bars) are presented. The identical statistical analysis was performed as in B.
We show that moss SPR2 is a MT minus end stabilising factor in protonemal cells, in which endoplasmic MTs are predominantly aligned along the cell’s long axis. Thus, minus end stabilisation is a conserved function of SPR2 in P. patens and Arabidopsis. It would be interesting to investigate if the stabilisation function is utilised also during cell division and in gametophore cells that organise cortical MT arrays.
We thank David Ehrhardt and Ram Dixit for communicating their results prior to publication; Fabien Nogue, Mitsuyasu Hasebe, Yuta Horiuchi, and Elena Kozgunova for plasmids and technical instructions; Rie Inaba and Momoko Nishina for technical assistance; Tetsuya Higashiyama for generous support on the project; This work was funded by the TORAY Science Foundation and JSPS KAKENHI 26711012 and 17H06471 (to G.G). M.Y. is a recipient of a JSPS pre-doctoral fellowship.
Conceived and designed the experiments: SY.L., M.Y., G.G. Performed the experiments and analysed the data: SY.L. Contributed reagents/materials: SY.L., M.Y., N.Y. Wrote the paper: SY.L., G.G.