Abstract
Phosphatidylserine (PS) is a constituent of the cell membrane, being especially abundant in the cytoplasmic leaflet, and plays important roles in a number of cellular functions, including the formation of cell polarity and intracellular vesicle transport. Several studies in mammalian cells have suggested the role of PS in retrograde membrane traffic through endosomes, but in yeast, where PS is localized primarily at the plasma membrane (PM), the role in intracellular organelles remains unclear. Additionally, it is reported that polarized endocytic site formation is defective in PS-depleted yeast cells, but the role in the endocytic machinery has not been well understood. In this study, to clarify the role of PS in the endocytic pathway, we analyzed the effect of PS depletion on endocytic internalization and post-endocytic transport. We demonstrated that in cell lacking the PS synthase Cho1p (cho1Δ cell), binding and internalization of mating pheromone α-factor into the cell was severely impaired. Interestingly, the processes of endocytosis were mostly unaffected, but protein transport from the trans-Golgi network (TGN) to the PM was defective and localization of cell surface proteins was severely impaired in cho1Δ cells. We also showed that PS accumulated in intracellular compartments in cells lacking Rcy1p and Vps52p, both of which are implicated in endosome-to-PM transport via the TGN, and that the number of Snx4p-residing endosomes was increased in cho1Δ cells. These results suggest that PS plays a crucial role in the transport and localization of cell surface membrane proteins.
Key words: phosphatidylserine, endocytosis, recycling, vesicle transport
Introduction
Phosphatidylserine (PS) is an anionic phospholipid present in the eukaryotic cell membrane, where it is a major constituent of the cytosolic leaflet. It is known to play important roles in various biological processes, including blood coagulation, phagocytosis of apoptotic cells, and recruitment of signaling molecules (Fadok et al., 1992; Lentz, 2003; Lucas and Cho, 2011). Additionally, PS dysregulation is known to be associated with different neurological diseases, such as Alzheimer’s disease and Parkinson’s disease (Ma et al., 2022). Certain viruses, such as Human Immunodeficiency Virus and Dengue Virus, are known to invade host cells through binding to PS in the outer leaflet of the plasma membrane (PM) (Moller-Tank and Maury, 2014). Therefore, to develop potential therapeutic targets for these diseases, it is important to clarify the cellular processes regulated by PS.
Previous studies have reported that PS is polarized at the PM and required for the development of cell polarity (Fairn et al., 2011). The highly conserved small GTPase, Cdc42, regulates the formation of cell polarity in eukaryotic cells (Etienne-Manneville, 2004; Pichaud et al., 2019). The yeast Saccharomyces cerevisiae forms polarized projections toward mating partners, bringing the cells into direct contact (Barral et al., 2000). Binding of the mating pheromone α-factor to the receptor Ste2p leads to recruitment of Cdc24, a guanine nucleotide exchange factor (GEF) for Cdc42p, and promotes Cdc42p activation at the tip of the mating projections, causing actin polymerization at presumptive bud sites (Butty et al., 2002; Wiget et al., 2004). In contrast, yeast mutants lacking the PS synthase, Cho1p (cho1Δ), show impairment of polarized Cdc42p localization, leading to a delay in bud emergence and defective mating (Fairn et al., 2011). Since the secretory pathway is required for the polarized distribution of PS (Fairn et al., 2011), intracellular vesicle transport seems to be important for the establishment of cell polarity, but the roles of PS in vesicle transport remain insufficiently understood. Additionally, it has not been clarified why PS depletion causes defective formation of mating projections.
Over the last decade, several probes, such as the C2 domain of lactadherin (Lact-C2) and the PH domain of evectin-2 (evt-2 PH), that bind to PS with high affinity have been developed, and studies using these probes have revealed that PS localizes to the PM and intracellular compartments in the endocytic pathway (Moravcevic et al., 2010; Uchida et al., 2011; Yeung et al., 2008). Sun and Drubin have reported the function of two anionic phospholipids, PI(4,5)P2 and PS, during clathrin-mediated endocytosis site initiation and vesicle formation in yeast (Sun and Drubin, 2012). They demonstrated that PI(4,5)P2 is essential for endocytic membrane invagination, whereas PS is important for the efficient recruitment and spatial restriction of endocytic proteins, such as the late clathrin coat protein Sla1p, to the PM (Sun and Drubin, 2012). Several studies in mammalian cells have revealed the role of PS in retrograde membrane traffic through endosomes (Kawasaki et al., 2022; Lee et al., 2015; Uchida et al., 2011). Uchida et al. have demonstrated that PS is highly concentrated in recycling endosomes (REs), thereby targeting evectin-2 to REs via its PH domain and controlling RE-to-Golgi transport (Uchida et al., 2011).
EHD1 (Eps15 homology domain-containing protein 1), a dynamin-like ATPase functioning in the endocytic pathway, is also shown to localize at endosomes by binding to PS (Lee et al., 2015; Naslavsky and Caplan, 2011). EHD1 is implicated in the retrograde transport mediated by SNX-BAR proteins, and knockdown of EHD1 causes defective recycling of endocytosed transferrin from REs to the PM (Lee et al., 2015; McKenzie et al., 2012). These observations suggest a specific role of PS at the endosomal compartment in mammalian cells. In yeast, PS was shown to distribute in various organelles, including the PM, Golgi, and vacuole (Tsuji et al., 2019), but it is still unclear whether PS has a specific role in endosomal compartments. Several studies have shown that type 4 P-type ATPases, which translocate PS from the extracellular to cytoplasmic leaflet of the cell membrane, are involved in endosome-to-Golgi transport (Furuta et al., 2007; Sakane et al., 2006), suggesting a role of PS in intracellular vesicle transport.
In the present study we show that in PS-deficient cho1Δ cells, binding and internalization of mating pheromone α-factor into the cell is impaired due to the decreased localization of receptor at the PM. We further demonstrate that localization of cell surface proteins at the PM is impaired in cho1Δ cells, and that these proteins are mis-sorted to the vacuole. We have also demonstrated that PS depletion causes an increase in the number of Snx4-residing endosomes. These findings suggest that PS plays a crucial role in the transport and localization of cell surface proteins to the PM.
Material and Methods
Yeast strains
The yeast strains used in this study are listed in Table S1. All strains were grown at 25°C in standard rich medium (YPD) or synthetic medium (SM) supplemented with 2% glucose and appropriate amino acids. The C-terminal fluorescent protein tagging of proteins was performed as described previously (Toshima et al., 2014)
Fluorescence microscopy and image analysis
Fluorescence microscopy was performed using an Olympus IX83 microscope equipped with a ×100/NA 1.40 (Olympus) or a ×100/NA 1.49 (Olympus) objective and Orca-R2 cooled CCD camera (Hamamatsu), using Metamorph software (Universal Imaging). Double-color imaging were performed using an Olympus IX81 microscope equipped with a high-speed filter changer (Lambda 10-3; Sutter Instruments) that can change filter sets within 40 ms. Simultaneous imaging of red and green fluorescence was performed using an Olympus IX83 microscope, described above, and an image splitter (Dual-View; Optical Insights) that divided the red and green components of the images with a 565-nm dichroic mirror and passed the red component through a 630/50 nm filter and the green component through a 530/30 nm filter. These split signals were taken simultaneously with one CCD camera, described above. All cells were imaged during the early- to mid-logarithmic phase. Images for analysis of co-localization of red and green signals were acquired using simultaneous imaging (64.5 nm pixel size), described above.
Fluorescence labeling of α-factor and endocytosis assays
Fluorescence labeling of α-factor was performed as described previously (Toshima et al., 2006). For endocytosis assays, cells were grown to an OD600 of ~0.5 in 0.5 ml YPD, briefly centrifuged, and resuspended in 20 μl SM with 5 μM Alexa Fluor 594-α-factor. After incubation on ice for 2 h, the cells were washed with ice-cold SM. Internalization was initiated by the addition of SM containing 4% glucose and amino acids at 25°C.
35S-labeled α-factor internalization and binding assays
Preparation and internalization of 35S-labeled α-factor was performed as described previously (Toshima et al., 2005). Briefly, cells were grown to an OD600 of ~0.3 in 50 ml YPD, briefly centrifuged and resuspended in 4 ml YPD containing 1% (w/v) BSA, 50 mM KH2PO4, pH 6, and 20 μg/ml of uracil, adenine, and histidine. After adding 35S-labeled α-factor, cell aliquots were withdrawn at various time points and subjected to a wash in pH 1.1 buffer to remove surface-bound α-factor so that internalized α-factor could be measured, or in pH 6 buffer to determine the total amount (internalized and bound) of α-factor. The amount of cell-associated radioactivity after each wash was determined by scintillation counting. Each experiment was performed at least three times. For the binding assay, cells grown to early to mid-logarithmic phase were briefly centrifuged, and resuspended in 50 μl SM with 1% (w/v) BSA and radiolabeled α-factor. After incubation on ice for 2 h, cells were washed with ice-cold SM and measured for their radioactivity.
Nanoluc luciferase-based secretion assay
Nanoluc (Nluc) luciferase reporter plasmid was expressed as follows: the Nluc fragment that contains signal sequence of MFA1 (nt 1-267) was amplified by PCR using pNL1.1 (Promega, Madison, WI) as a template, and subcloned into the HindIII- and XhoI-digested pBlueScript II SK (pBS-Nluc), and the SacI-HindIII fragment of the 417-bp 5' UTR of TPI1 gene, which contains the S. cerevisiae TPI1 promoter, was amplified by PCR using yeast genome DNA as a template, and inserted into SacI- and HindIII-digested pBS-Nluc (pBS-PTPI1-Nluc). Next, the XhoI-ApaI fragment of the 200-bp 3' UTR of TPI1 gene, which contains the S. cerevisiae TPI1 terminator, was inserted into XhoI- and ApaI-digested pBS-PTPI1-Nluc (pBS-PTPI-Nluc-TTPI). To create an integration plasmid, the SacI-ApaI fragment of the PTPI-Nluc-TTPI was inserted into the SacI- and ApaI-digested pRS305 (pRS305-PTPI-Nluc-TTPI). To integrate pRS305-PTPI-Nluc-TTPI at the LEU2 locus, the plasmid was linearized by EcoRI and transformed into wild-type or mutant cells. For the secretion assay, cells expressing Nluc luciferase reporter were grown to an OD600 of ~0.5 in 1.0 ml YPD, briefly centrifuged, and resuspended in fresh 800 μl YPD. At each time point, 100 μl of the culture medium was aliquoted and centrifuged, and luciferase activities in the supernatants were measured by Nano-Glo luciferase assay system (Promega, Madison, WI).
Results
PS is required for cell surface localization of the Ste2 receptor
A previous study has demonstrated that PS is required for proper Cdc42 localization and establishment of cell polarity, and that PS depletion causes depolarization of Cdc42p, leading to a delay in bud emergence and, subsequently, defective mating (Fairn et al., 2011). First, therefore, we confirmed that whether PS synthesis is required for the morphological change (shmoo morphology) in response to the mating pheromone α-factor. At 2 h after α-factor treatment, ~82% of wild-type cell exhibited shmoo morphology, whereas cho1Δ cells rarely did so (~4%) (Fig. 1A). To examine whether cho1Δ cells have defects in α-factor binding or in α-factor-induced signal transduction, we next examined the binding and uptake of α-factor into the cells. We labeled wild-type and cho1Δ cells with Alexa Fluor 594-conjugated α-factor (A594-α-factor) and observed the localization. We found that the fluorescence intensity of A594-α-factor bound to cho1Δ cells was decreased relative to wild-type cells (Fig. 1B). In contrast, A594-α-factor binding was barely observed in cells lacking the α-factor receptor Ste2p (Fig. 1C), confirming that α-factor only marginally binds to cho1Δ cells (Fig. 1C). We also observed an appreciable delay of A594-α-factor internalization in cho1Δ cells (Fig. 1D). Quantitative analysis categorizing A594-α-factor localization as PM only, PM and endosome, or endosome and vacuole revealed that cho1Δ cells had obvious defects in α-factor internalization (Fig. 1E). To examine in detail the effect of PS depletion on α-factor binding and uptake into the cell, we compared them using 35S-labeled α-factor. Similarly to A594-α-factor, 35S-labeled α-factor was efficiently incorporated into wild-type cells, whereas it was incorporated only slightly into cho1Δ cells (Fig. 1F). Surprisingly, binding of α-factor to cho1Δ cells was decreased to ~10% in comparison to wild-type cells, and the amount of binding was almost the same as that to ste2Δ cells (Fig. 1G). As A594-α-factor slightly bound to cho1Δ cells while 35S-labeled α-factor little bound, we speculated that changes in lipid composition in the PM by PS depletion might cause non-specific binding of A594-α-factor to the cell. As expected, we found that A594-α-factor barely binds to ste2Δ cells but binds to ste2Δ cells lacking Cho1p at the similar level to cho1Δ cells (Fig. 1B, H), suggesting that A594-α-factor binds to cho1Δ cells independently of Ste2 receptor. Additionally, we found that in cho1Δ cells Ste2p localization was markedly shifted to intracellular compartments, in contrast to its association with the PM in wild-type cells (Fig. 1I). While most wild-type cells obviously exhibited Ste2-GFP at the PM as well as the vacuole, cho1Δ cells showed little PM localization of Ste2p (Fig. 1I, J). Intracellular Ste2-GFP was highly colocalized with mCherry-fused Pep4p in both wild-type and cho1Δ cells (Fig. 1I), indicating that PS depletion leads to mis-sorting of Ste2p from the PM to the vacuole.
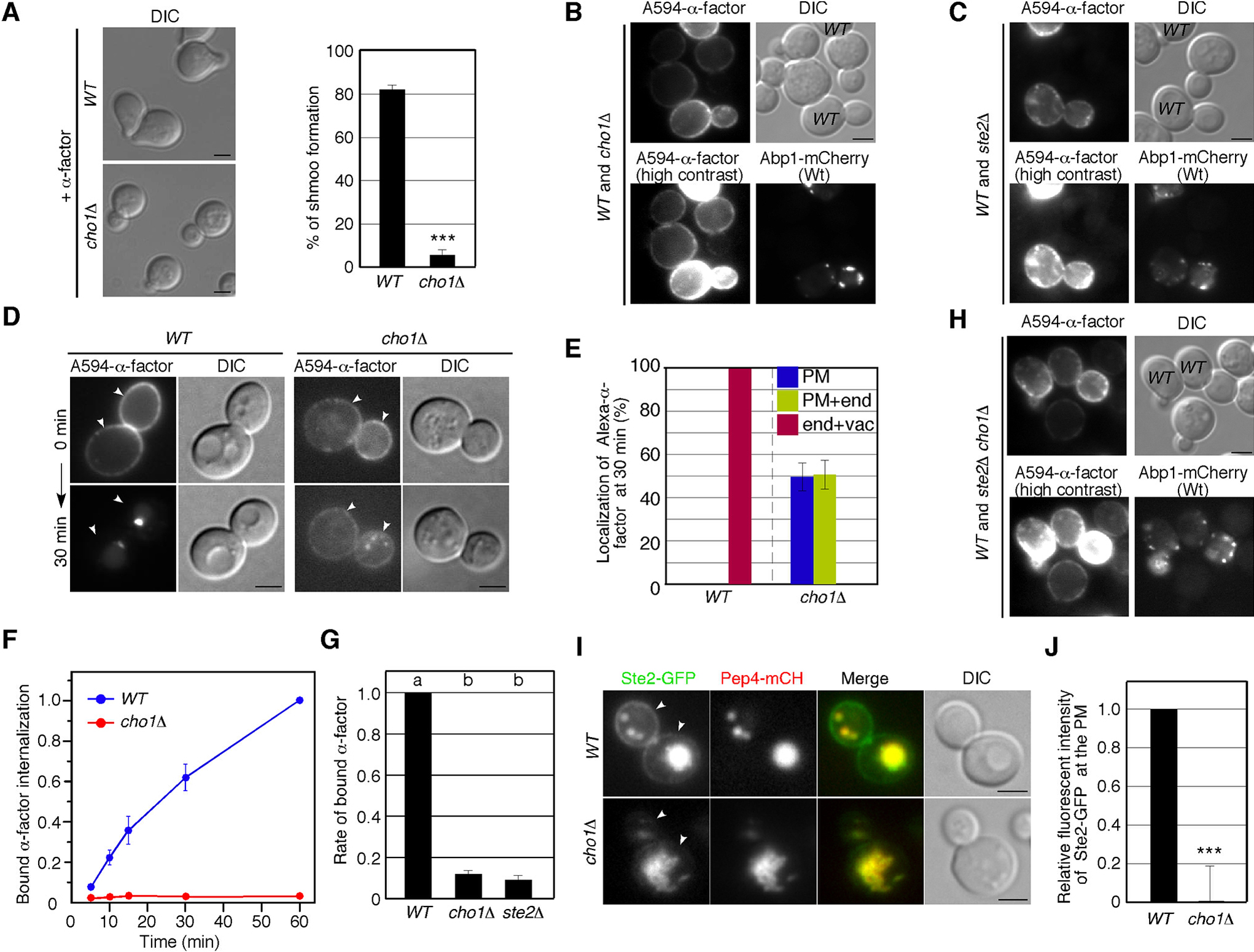
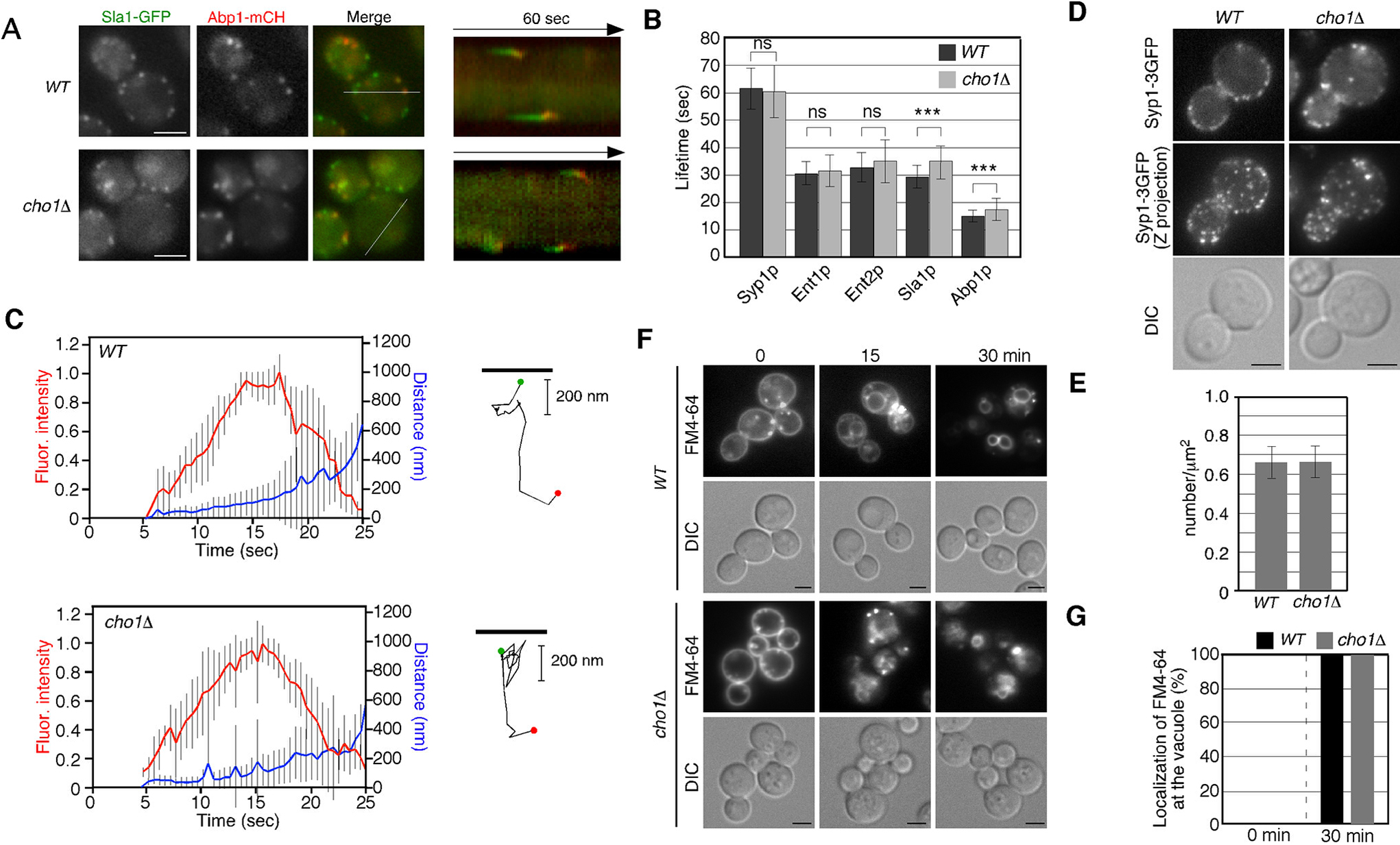
Given the reduced binding and internalization of fluorescent α-factor to the PM, we next investigated the effect of CHO1 gene deletion on the formation and internalization of endocytic vesicles. We used Sla1-GFP and Abp1-mRFP as markers to follow the dynamics of endocytic vesicles (Fig. 2A) (Kaksonen et al., 2003). First, we carried out simultaneous imaging of Sla1p and Abp1p to analyze the dynamic behavior of these proteins in live cells. In wild-type cells, Sla1-GFP was immediately followed by Abp1-mRFP recruitment, as shown in kymographs generated across a single pixel-wide line for an individual patch (Fig. 2A). Similar dynamics of Sla1-GFP and Abp1-mRFP patches were observed in cho1Δ cells (Fig. 2A), although the lifetime of Sla1-GFP was slightly prolonged in cho1Δ cells relative to wild-type cells (~29.4 vs. ~35.1 sec). This result is consistent with previous observations (Sun and Drubin, 2012). We next examined the effect of CHO1 gene deletion on the dynamics of endocytic vesicles. Fluorescence intensity and particle tracking analysis showed that Abp1p patches in both wild-type and cho1Δ cells were internalized normally (Fig. 2C), although the lifetime of Abp1-mCherry in the cho1Δ cells was also slightly prolonged in comparison with that in wild-type cells (~15.1 vs. ~18.4 sec) (Fig. 2B). In contrast, the lifetimes of the early coat protein, Syp1p, and the mid coat proteins, Ent1p/2p, were almost the same in both wild-type and cho1Δ cells (Fig. 2B). To further examine the effect of PS depletion on clathrin coat assembly, we assessed the number of endocytic sites in cho1Δ cells using Syp1-3GFP (Suzuki et al., 2012). Maximum-intensity Z-projections of live cells were analyzed to determine the average number of patches per unit surface area, and patch densities were quantified only in mother cells, where individual patches were distinguishable. As shown in Fig. 2D, in cho1Δ cells, there was no change in the number of Syp1p patches per unit surface area relative to wild-type cells. These results clearly demonstrated that endocytic vesicle formation and internalization occur normally in cho1Δ cells. Finally, we examined the internalization of 3-triethylammoniumpropyl-4-p-diethylaminophenylhexatrienyl pyridinium dibromide (FM4-64), a lipophilic styryl dye that is used to label bulk endocytosis. When added to wild-type cells, FM4-64 is immediately incorporated into the PM, internalized via bulk endocytosis, and then transported to the vacuole within 30 min (Fig. 2F, G). This assay revealed that cho1Δ cells showed little delay in membrane internalization, suggesting that the endocytic machinery is not impaired in the cho1Δ cell.
To clarify whether the decreased localization of Ste2p at the PM in cho1Δ cells is caused by a recycling defect, we examined the effect of cho1Δ on trafficking of GFP-Snc1p, an exocytic v-SNARE that is endocytosed, transiently localized to early endosomes, and recycled back to the PM via the trans-Golgi network (TGN) (Galan et al., 2001; Lewis et al., 2000). It has been shown that mutations affecting endosome-mediated trafficking often cause mis-localization of Snc1p from the PM to the endosomal or vacuolar compartments (Galan et al., 2001; Lewis et al., 2000; Reggiori et al., 2003). In wild-type cells, GFP-Snc1p was localized at the PM with some punctate staining of internal structures (Fig. 3A), as shown in previous studies (Lewis et al., 2000). Intriguingly, in cho1Δ cells, localization of GFP-Snc1 was shifted to intracellular compartments (Fig. 3A). Quantitative analysis showed that the fluorescence intensity of GFP-Snc1 at the PM was decreased to ~9% in cho1Δ cells relative to that in wild-type cells (Fig. 3B). These intracellular structures were significantly co-localized with Pep4-mCherry, a marker for the vacuole (Fig. 3A). To examine whether PS synthesis is generally required for the localization of cell surface protein at the PM, we used the yeast arginine permease Can1p, which is known to be recycled to the PM via the TGN after endocytic internalization (Gournas et al., 2017). Similarly to Ste2p, most wild-type cells exhibited localization of Can1-GFP at the PM, but the cell surface localization was markedly decreased to ~1.3% in cho1Δ cells (Fig. 3C, D). We also examined the localization of the cell wall sensor protein Wsc1p, which is maintained by endocytosis and recycling from endosomes back to the cell surface (Piao et al., 2007; Ueno et al., 2014). While GFP-tagged Wsc1p is localized primarily at the PM in wild-type cells, it was localized at the vacuole in cho1Δ cells (Fig. 3E), suggesting that PS is generally required for the localization of cell surface protein at the PM. Quantitative analysis showed that only ~2.7% of wild-type cells had Wsc1-GFP at the vacuole, whereas ~90.7% of cho1Δ cells exhibited vacuolar localization (Fig. 3F). To further characterize the GFP-Snc1 localization defect in cho1Δ cells, we used a GFP-fused Snc1(en-) mutant containing two mutations, V40A and M43A, that interfere with endocytosis and cause GFP-Snc1 to accumulate on the PM in wild-type cells (Fig. 3G) (Lewis et al., 2000). We observed that GFP-Snc1(en-) was localized to the vacuole in addition to the PM in cho1Δ cells, suggesting defective transport from the TGN to the PM (Fig. 3G, H). Consistent with this, the fluorescence intensity of GFP-Snc1(en-) at the PM was decreased to ~75.6% in cho1Δ cells relative to that in wild-type cells (Fig. 3I). To further confirm this, we examined the effect on the secretory pathway using NanoLuc (Nluc) luciferase reporter, which contains a secretory signal for α-factor at the N terminus. The Nluc luciferase reporter gene was chromosomally integrated in both wild-type and cho1Δ cells, and the luciferase activity in the culture medium was assessed in both cases. Little luciferase activity was detected in the culture supernatant of wild-type cells, which did not express Nluc luciferase (mock), whereas the activity increased steadily until the end of measurement in cells expressing Nluc luciferase (Fig. 3J). Interestingly, the luciferase activity in the culture supernatant of cho1Δ cells was significantly decreased while the intracellular activity was increased (Fig. 3J, K). Additionally, we examined the growth of cho1Δ cells on media containing sucrose as a sole carbon source, because secretion of invertase, encoded by SUC2, is prerequisite for yeast cell growth on the media (Fig. 3L) (Novick et al., 1980). Consistent with the result obtained using the NLuc luciferase reporter, the cho1Δ cells were unable to grow on media containing sucrose (Fig. 3L). These results, taken together, suggested that cho1Δ cells have a defect at least in the pathway from the TGN to the PM, and that cell surface proteins recycled back to the PM might be mis-sorted to the vacuole at the TGN.

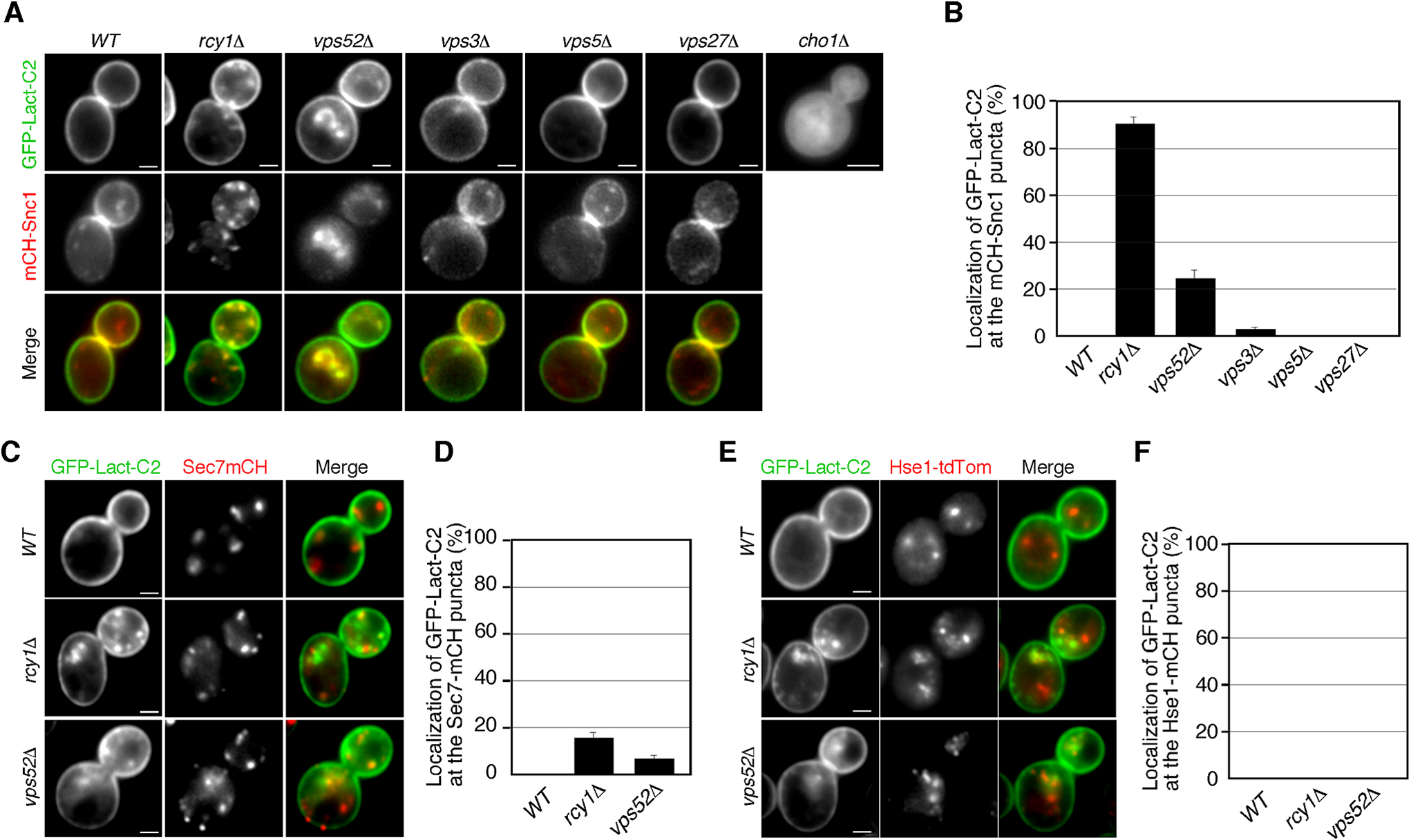
To evaluate the potential function of PS in cell surface protein recycling, we next investigated its subcellular localization using the stereospecific PS-binding C2 domain of bovine lactadherin (Lact-C2) (Leventis and Grinstein, 2010; Yeung et al., 2008). First, we introduced a fluorescent PS-specific probe, GFP-fused Lact-C2 (GFP-Lact-C2), to wild-type and cho1Δ cells and observed its localization. As shown previously, in wild-type cells, Lact-C2-GFP was localized exclusively at the PM, whereas it was largely cytoplasmic in cho1Δ cells (Fig. 4A) (Fairn et al., 2011; Sun and Drubin, 2012). As described above, as cho1Δ cells have severe defects in the TGN-to-PM pathway but show normal endocytic internalization, we speculated that PS might transiently localize at intracellular compartments, such as the TGN and endosomes, and regulate the recycling pathway. Previous studies have shown that in cells lacking Rcy1p, an F-box protein involved in the recycling of cell surface proteins, Lact-C2-GFP accumulates at intracellular membrane compartments and co-localizes with mRFP-Snc1 there (Galan et al., 2001; Ma and Burd, 2020; Wiederkehr et al., 2000). As reported previously, we observed that GFP-Lact-C2 was highly co-localized with mCherry-Snc1p at intracellular puncta in rcy1Δ cells (~90.0%) (Fig. 4B, C). Previous studies have also demonstrated that in rcy1Δ cells Snc1p partially co-localizes with Tlg1p at the early endosome, but does not co-localize with Sec7p, which is localized at the TGN (Best et al., 2020; Ma and Burd, 2019). Similarly to the case of Snc1p and Sec7p, GFP-Lact-C2 was not well co-localized with Sec7p-residing TGN (~14.7%) in the rcy1Δ mutant (Fig. 4C, D). We also compared the PS localization with Hse1-tdTomato, a marker of endosomal compartments, but no co-localization was observed (Fig. 4E, F). These results suggest that neither the TGN nor endosome is the region that PS accumulates. To further examine possible intracellular compartments at which PS is localized, we utilized three other mutants with defects in the intracellular vesicle trafficking pathway. Among them, we found that Lact-C2-GFP also accumulates at intracellular membrane compartments in vps52Δ cells. The VPS52 gene encodes a subunit of the GARP (Golgi-associated retrograde protein) complex, which is a protein complex involved in the recycling of proteins from endosomes to the TGN (Bonifacino and Hierro, 2011). In about 24% of the vps52Δ mutants, GFP-Lact-C2 was localized at intracellular puncta where Snc1p resides, but Sec7p and Hse1p do not (Fig. 4B–F). This indicated that transport of PS from the endosome to the TGN is partially impaired in the vps52Δ mutant. In contrast, GFP-Lact-C2 exhibited PM localization, similar to that in wild-type cells, in cells lacking the VPS3, VPS5 or VPS27 gene, which encode proteins required for the early-to-late endosome, late endosome-to-TGN, or late endosome-to-vacuole trafficking (Fig. 4A) (Bowers and Stevens, 2005). These observations suggest a possibility that PS is endocytosed and recycled back to the PM through the endosome and the TGN compartments.
Since PS appears to localize transiently at the TGN and endosome, we next examined the effects of PS depletion on these compartments. First, we examined the localization of Kex2, which is known to localize to both the TGN and endosomal compartments. Kex2p is a pheromone-processing protease residing at the TGN, being transported to the endosome and then recycled back to the TGN in a retromer-dependent manner (Lewis et al., 2000; Seaman et al., 1998). In cho1Δ cells the number of Sec7p-residing TGN was almost the same as that in wild-type cells (Fig. 5A, B), but the fluorescence intensity of Kex2-GFP at the vacuole was significantly increased (Fig. 5C). These observations indicated that the retrieval pathway from the endosome to the TGN is impaired in cho1Δ cells. Next, to determine the effect on endosomes in cho1Δ cells, we examined the localization of Snx4p, which is a subunit of the sorting nexin complex that mediates a retrieval pathway from the endosome to the TGN (Hettema et al., 2003). GFP-Snx4 puncta were well co-localized with Hse1-tdTomato-residing endosomes in both wild-type and cho1Δ cells (Fig. 5D). Intriguingly, we found that the number of these GFP-Snx4-residing puncta was significantly increased in cho1Δ cells (Fig. 5D, E). These observations suggest that PS depletion affects transport from the endosome to the TGN, resulting in an increase in the number of endosomal compartments.

Discussion
In this study, we have shown that PS synthesis is important for the localization of cell surface proteins. A previous study had demonstrated that cells lacking CHO1 gene have significantly decreased mating efficiency due to impairment of the polarity of Cdc42p, which is required for the formation of mating projections (Fairn et al., 2011). In the study it had also shown that PS depletion in cho1Δ cell causes a decrease in Phosphatidylehanolamine (PE) content, but Cdc42 accumulation correlates with the presence and polarization of PS, and not changes in PE content (Fairn et al., 2011). In the present study, we demonstrated that cho1Δ cells have defect in the localization of cell surface proteins at the PM. Thus, it seems that the decreased mating efficiency observed in cho1Δ cells is caused by defective transport of Ste2p from the TGN to the PM (Fig. 5F, G). While it is known that binding to α-factor triggers Ste2p transport through the endocytic pathway to the vacuole (Hicke and Riezman, 1996; Toshima et al., 2009), it is still unclear whether Ste2p in the absence of α-factor is recycled back to the PM. In our previous study, we showed that cells lacking the Arf-GAP Glo3p exhibit a dramatic reduction of α-factor binding to the cell surface (Kawada et al., 2015). The glo3Δ mutant had prominent defects in endosome-to-TGN trafficking, suggesting that Ste2p cannot be efficiently recycled back to the PM (Kawada et al., 2015). In cho1Δ cells, several cell surface proteins, including Ste2p, were mis-transported to the vacuole. We also showed that the GFP-Snc1(en-) mutant, which is not endocytosed, was localized to both the PM and the vacuole, indicating that TGN-to-PM transport was not completely disrupted. These observations suggest that defects in both the secretion and recycling pathways would decrease the localization of Ste2p at the PM in cho1Δ cells (Fig. 5G). In wild-type cells, we propose that Ste2p is also endocytosed constitutively, but then retrieved to the TGN from the endosomal compartment and shuttled again back to the cell surface (Fig. 5F).
Several studies have reported that PS is required for intracellular vesicle transport and that cho1Δ cells exhibit defects in endocytic site formation (Sun and Drubin, 2012). It has also been shown that in cho1Δ cells Sla1p does not preferentially localize in the small buds where endocytic actin patches are normally concentrated (Sun and Drubin, 2012). Similarly to these previous observations, we have shown that most cho1Δ cells lose their polarized localization of endocytic sites. However, the localization, lifetime, and amount of Syp1p, which localizes at the earliest stage of endocytosis, appear to be mostly normal, suggesting that endocytic site formation in the mutant is not severely affected. Additionally, actin patch dynamics were almost equivalent in both wild-type and cho1Δ cells. Thus, it remains unclear why the lifetimes of Sla1p and Abp1p are prolonged in cho1Δ cells, and further studies to determine the role of PS in the endocytic pathway will be needed.
In mammalian cells, PS is known to be important for retrograde membrane traffic at recycling endosomes (Uchida et al., 2011). PS localizes preferentially at recycling endosomes, and masking of intracellular PS by overexpression of Lact-C2 suppressed membrane traffic from REs to the Golgi (Uchida et al., 2011). In the process of retrograde trafficking, a dynamin-like ATPase EHD1 has been shown to be recruited to endosomes through its PS-binding ability (Lee et al., 2015). EHD1 facilitates the fission of tubulovesicular carriers from endosomes, and a reduction of PS decreases the localization of EHD1 at endosomes containing SNX1, a component of the SNX1-BAR complex, resulting in the fission defect (Deo et al., 2018; Kamerkar et al., 2019; Kawasaki et al., 2022). In yeast, it has demonstrated that the SNX-BAR protein, Snx4-Atg20 dimer, also preferentially binds and remodels the PS-containing membrane, thereby exporting the PS-rich membrane from the endosome to promote autophagy and vacuole membrane fusion (Ma et al., 2018). Additionally, a previous study reported that PS flipping to the cytosolic leaflet of endosomal membrane by yeast Drs2p, a phospholipid translocase in the type IV P-type ATPase family, is crucial for generating membrane curvature at the endosomes (Xu et al., 2013). This Drs2p flippase activity is required for Gcs1 endosomal localization via its +ALPS motif and protein transport between early endosome and the TGN (Xu et al., 2013), suggesting a role of PS in the vesicle transport between these organelles. Our present findings also suggest the general importance of PS in the recycling of cell surface proteins. In cells lacking Rcy1p or Vps52p, we have shown that PS accumulates at intracellular membrane compartments that mediate endosome-to-Golgi trafficking. In contrast, in cells lacking Vps3p, Vps5p, or Vps27p, PS localization appears to be normal. Vps3p is a subunit of the CORVET complex involved in early-to-late endosomal maturation (Nordmann et al., 2010), and Vps5p, a subunit of the retromer complex, mediates transport from the prevacuolar endosomal compartment to the TGN (Nothwehr et al., 2000; Seaman, 2007). Vps27p recruits the ESCRT machinery to endosomes during multivesicular body (MVB) sorting (Katzmann et al., 2003). Deletion of these proteins is known to cause mis-sorting of cargos to abnormal endosome structures or vacuoles (Bowers and Stevens, 2005). Thus, the normal localization of Lact-C2 to the PM suggests that PS is localized at the early-stage endosome, and not transported to the late endosomal compartment. This idea is consistent with a previous observation that retrograde sorting of PS from the early endosome by Snx4 family sorting nexins is important for ensuring that vacuolar fusion occurs at a later stage (Ma et al., 2018). Our finding that the number of endosomes containing Snx4p was increased in cho1Δ cells also supports this idea.
Conflict of Interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by JSPS KAKENHI GRANT #18K062291, and the Takeda Science Foundation to J.Y.T., as well as JSPS KAKENHI GRANT #19K065710, the Takeda Science Foundation, and Life Science Foundation of JAPAN to J.T.
References
- Barral, Y., Mermall, V., Mooseker, M.S., and Snyder, M. 2000. Compartmentalization of the cell cortex by septins is required for maintenance of cell polarity in yeast. Mol. Cell, 5: 841–851.
- Best, J.T., Xu, P., McGuire, J.G., Leahy, S.N., and Graham, T.R. 2020. Yeast synaptobrevin, Snc1, engages distinct routes of postendocytic recycling mediated by a sorting nexin, Rcy1-COPI, and retromer. Mol. Biol. Cell, 31: 944–962.
- Bonifacino, J.S. and Hierro, A. 2011. Transport according to GARP: receiving retrograde cargo at the trans-Golgi network. Trends Cell Biol., 21: 159–167.
- Bowers, K. and Stevens, T.H. 2005. Protein transport from the late Golgi to the vacuole in the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta, 1744: 438–454.
- Butty, A.C., Perrinjaquet, N., Petit, A., Jaquenoud, M., Segall, J.E., Hofmann, K., Zwahlen, C., and Peter, M. 2002. A positive feedback loop stabilizes the guanine-nucleotide exchange factor Cdc24 at sites of polarization. EMBO J., 21: 1565–1576.
- Deo, R., Kushwah, M.S., Kamerkar, S.C., Kadam, N.Y., Dar, S., Babu, K., Srivastava, A., and Pucadyil, T.J. 2018. ATP-dependent membrane remodeling links EHD1 functions to endocytic recycling. Nat. Commun., 9: 5187.
- Etienne-Manneville, S. 2004. Cdc42—the centre of polarity. J. Cell Sci., 117: 1291–1300.
- Fadok, V.A., Voelker, D.R., Campbell, P.A., Cohen, J.J., Bratton, D.L., and Henson, P.M. 1992. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol., 148: 2207–2216.
- Fairn, G.D., Hermansson, M., Somerharju, P., and Grinstein, S. 2011. Phosphatidylserine is polarized and required for proper Cdc42 localization and for development of cell polarity. Nat. Cell Biol., 13: 1424–1430.
- Furuta, N., Fujimura-Kamada, K., Saito, K., Yamamoto, T., and Tanaka, K. 2007. Endocytic recycling in yeast is regulated by putative phospholipid translocases and the Ypt31p/32p-Rcy1p pathway. Mol. Biol. Cell, 18: 295–312.
- Galan, J.M., Wiederkehr, A., Seol, J.H., Haguenauer-Tsapis, R., Deshaies, R.J., Riezman, H., and Peter, M. 2001. Skp1p and the F-box protein Rcy1p form a non-SCF complex involved in recycling of the SNARE Snc1p in yeast. Mol. Cell. Biol., 21: 3105–3117.
- Gournas, C., Saliba, E., Krammer, E.M., Barthelemy, C., Prévost, M., and André, B. 2017. Transition of yeast Can1 transporter to the inward-facing state unveils an α-arrestin target sequence promoting its ubiquitylation and endocytosis. Mol. Biol. Cell, 28: 2819–2832.
- Hettema, E.H., Lewis, M.J., Black, M.W., and Pelham, H.R. 2003. Retromer and the sorting nexins Snx4/41/42 mediate distinct retrieval pathways from yeast endosomes. EMBO J., 22: 548–557.
- Hicke, L. and Riezman, H. 1996. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell, 84: 277–287.
- Kaksonen, M., Sun, Y., and Drubin, D.G. 2003. A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell, 115: 475–487.
- Kamerkar, S.C., Roy, K., Bhattacharyya, S., and Pucadyil, T.J. 2019. A Screen for Membrane Fission Catalysts Identifies the ATPase EHD1. Biochemistry, 58: 65–71.
- Katzmann, D.J., Stefan, C.J., Babst, M., and Emr, S.D. 2003. Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J. Cell Biol., 162: 413–423.
- Kawada, D., Kobayashi, H., Tomita, T., Nakata, E., Nagano, M., Siekhaus, D.E., Toshima, J.Y., and Toshima, J. 2015. The yeast Arf-GAP Glo3p is required for the endocytic recycling of cell surface proteins. Biochim. Biophys. Acta, 1853: 144–156.
- Kawasaki, A., Sakai, A., Nakanishi, H., Hasegawa, J., Taguchi, T., Sasaki, J., Arai, H., Sasaki, T., Igarashi, M., and Nakatsu, F. 2022. PI4P/PS countertransport by ORP10 at ER-endosome membrane contact sites regulates endosome fission. J. Cell Biol., 221.
- Lee, S., Uchida, Y., Wang, J., Matsudaira, T., Nakagawa, T., Kishimoto, T., Mukai, K., Inaba, T., Kobayashi, T., Molday, R.S., Taguchi, T., and Arai, H. 2015. Transport through recycling endosomes requires EHD1 recruitment by a phosphatidylserine translocase. EMBO J., 34: 669–688.
- Lentz, B.R. 2003. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog. Lipid Res., 42: 423–438.
- Leventis, P.A. and Grinstein, S. 2010. The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys., 39: 407–427.
- Lewis, M.J., Nichols, B.J., Prescianotto-Baschong, C., Riezman, H., and Pelham, H.R. 2000. Specific retrieval of the exocytic SNARE Snc1p from early yeast endosomes. Mol. Biol. Cell, 11: 23–38.
- Lucas, N. and Cho, W. 2011. Phosphatidylserine binding is essential for plasma membrane recruitment and signaling function of 3-phosphoinositide-dependent kinase-1. J. Biol. Chem., 286: 41265–41272.
- Ma, M., Kumar, S., Purushothaman, L., Babst, M., Ungermann, C., Chi, R.J., and Burd, C.G. 2018. Lipid trafficking by yeast Snx4 family SNX-BAR proteins promotes autophagy and vacuole membrane fusion. Mol. Biol. Cell, 29: 2190–2200.
- Ma, M.X. and Burd, C.G. 2019. Retrograde trafficking and quality control of yeast synaptobrevin, Snc1, are conferred by its transmembrane domain. Mol. Biol. Cell, 30: 1729–1742.
- Ma, M.X. and Burd, C.G. 2020. Retrograde trafficking and plasma membrane recycling pathways of the budding yeast Saccharomyces cerevisiae. Traffic, 21: 45–59.
- Ma, X., Li, X., Wang, W., Zhang, M., Yang, B., and Miao, Z. 2022. Phosphatidylserine, inflammation, and central nervous system diseases. Front. Aging Neurosci., 14: 975176.
- McKenzie, J.E., Raisley, B., Zhou, X., Naslavsky, N., Taguchi, T., Caplan, S., and Sheff, D. 2012. Retromer guides STxB and CD8-M6PR from early to recycling endosomes, EHD1 guides STxB from recycling endosome to Golgi. Traffic, 13: 1140–1159.
- Moller-Tank, S. and Maury, W. 2014. Phosphatidylserine receptors: enhancers of enveloped virus entry and infection. Virology, 468–470: 565–580.
- Moravcevic, K., Mendrola, J.M., Schmitz, K.R., Wang, Y.H., Slochower, D., Janmey, P.A., and Lemmon, M.A. 2010. Kinase associated-1 domains drive MARK/PAR1 kinases to membrane targets by binding acidic phospholipids. Cell, 143: 966–977.
- Naslavsky, N. and Caplan, S. 2011. EHD proteins: key conductors of endocytic transport. Trends Cell Biol., 21: 122–131.
- Nordmann, M., Cabrera, M., Perz, A., Brocker, C., Ostrowicz, C., Engelbrecht-Vandre, S., and Ungermann, C. 2010. The Mon1-Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7. Curr. Biol., 20: 1654–1659.
- Nothwehr, S.F., Ha, S.A., and Bruinsma, P. 2000. Sorting of yeast membrane proteins into an endosome-to-Golgi pathway involves direct interaction of their cytosolic domains with Vps35p. J. Cell Biol., 151: 297–310.
- Novick, P., Field, C., and Schekman, R. 1980. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell, 21: 205–215.
- Piao, H.L., Machado, I.M., and Payne, G.S. 2007. NPFXD-mediated endocytosis is required for polarity and function of a yeast cell wall stress sensor. Mol. Biol. Cell, 18: 57–65.
- Pichaud, F., Walther, R.F., and Nunes de Almeida, F. 2019. Regulation of Cdc42 and its effectors in epithelial morphogenesis. J. Cell Sci., 132.
- Reggiori, F., Wang, C.W., Stromhaug, P.E., Shintani, T., and Klionsky, D.J. 2003. Vps51 is part of the yeast Vps fifty-three tethering complex essential for retrograde traffic from the early endosome and Cvt vesicle completion. J. Biol. Chem., 278: 5009–5020.
- Sakane, H., Yamamoto, T., and Tanaka, K. 2006. The functional relationship between the Cdc50p-Drs2p putative aminophospholipid translocase and the Arf GAP Gcs1p in vesicle formation in the retrieval pathway from yeast early endosomes to the TGN. Cell Struct. Funct., 31: 87–108.
- Seaman, M.N. 2007. Identification of a novel conserved sorting motif required for retromer-mediated endosome-to-TGN retrieval. J. Cell Sci., 120: 2378–2389.
- Seaman, M.N., McCaffery, J.M., and Emr, S.D. 1998. A membrane coat complex essential for endosome-to-Golgi retrograde transport in yeast. J. Cell Biol., 142: 665–681.
- Sun, Y. and Drubin, D.G. 2012. The functions of anionic phospholipids during clathrin-mediated endocytosis site initiation and vesicle formation. J. Cell Sci., 125: 6157–6165.
- Suzuki, R., Toshima, J.Y., and Toshima, J. 2012. Regulation of clathrin coat assembly by Eps15 homology domain-mediated interactions during endocytosis. Mol. Biol. Cell, 23: 687–700.
- Toshima, J., Toshima, J.Y., Martin, A.C., and Drubin, D.G. 2005. Phosphoregulation of Arp2/3-dependent actin assembly during receptor-mediated endocytosis. Nat. Cell Biol., 7: 246–254.
- Toshima, J.Y., Toshima, J., Kaksonen, M., Martin, A.C., King, D.S., and Drubin, D.G. 2006. Spatial dynamics of receptor-mediated endocytic trafficking in budding yeast revealed by using fluorescent alpha-factor derivatives. Proc. Natl. Acad. Sci., 103: 5793–5798.
- Toshima, J.Y., Nakanishi, J.I., Mizuno, K., Toshima, J., and Drubin, D.G. 2009. Requirements for Recruitment of a G Protein-coupled Receptor to Clathrin-coated Pits in Budding Yeast. Mol. Biol. Cell, 20: 5039–5050.
- Toshima, J.Y., Nishinoaki, S., Sato, Y., Yamamoto, W., Furukawa, D., Siekhaus, D.E., Sawaguchi, A., and Toshima, J. 2014. Bifurcation of the endocytic pathway into Rab5-dependent and -independent transport to the vacuole. Nat. Commun., 5: 3498.
- Tsuji, T., Cheng, J., Tatematsu, T., Ebata, A., Kamikawa, H., Fujita, A., Gyobu, S., Segawa, K., Arai, H., Taguchi, T., Nagata, S., and Fujimoto, T. 2019. Predominant localization of phosphatidylserine at the cytoplasmic leaflet of the ER, and its TMEM16K-dependent redistribution. Proc. Natl. Acad. Sci., 116: 13368–13373.
- Uchida, Y., Hasegawa, J., Chinnapen, D., Inoue, T., Okazaki, S., Kato, R., Wakatsuki, S., Misaki, R., Koike, M., Uchiyama, Y., Iemura, S., Natsume, T., Kuwahara, R., Nakagawa, T., Nishikawa, K., Mukai, K., Miyoshi, E., Taniguchi, N., Sheff, D., Lencer, W.I., Taguchi, T., and Arai, H. 2011. Intracellular phosphatidylserine is essential for retrograde membrane traffic through endosomes. Proc. Natl. Acad. Sci., 108: 15846–15851.
- Ueno, K., Saito, M., Nagashima, M., Kojima, A., Nishinoaki, S., Toshima, J.Y., and Toshima, J. 2014. V-ATPase-dependent luminal acidification is required for endocytic recycling of a yeast cell wall stress sensor, Wsc1p. Biochem. Biophys. Res. Commun., 443: 549–555.
- Wiederkehr, A., Avaro, S., Prescianotto-Baschong, C., Haguenauer-Tsapis, R., and Riezman, H. 2000. The F-box protein Rcy1p is involved in endocytic membrane traffic and recycling out of an early endosome in Saccharomyces cerevisiae. J. Cell Biol., 149: 397–410.
- Wiget, P., Shimada, Y., Butty, A.C., Bi, E., and Peter, M. 2004. Site-specific regulation of the GEF Cdc24p by the scaffold protein Far1p during yeast mating. EMBO J., 23: 1063–1074.
- Xu, P., Baldridge, R.D., Chi, R.J., Burd, C.G., and Graham, T.R. 2013. Phosphatidylserine flipping enhances membrane curvature and negative charge required for vesicular transport. J. Cell Biol., 202: 875–886.
- Yeung, T., Gilbert, G.E., Shi, J., Silvius, J., Kapus, A., and Grinstein, S. 2008. Membrane phosphatidylserine regulates surface charge and protein localization. Science, 319: 210–213.
