Abstract
The current worldwide energy problems resulting from global environmental issues require highly efficient, environmentally benign energy storage technologies. Among various innovative battery types, rechargeable batteries based on magnesium (Mg) metal anodes represent an attractive option because of their large volumetric/gravimetric capacities and the cost-effectiveness of the base Mg metal. To realize practical Mg batteries, intense efforts have been focused on the development of cathode and electrolyte materials. Defect spinel oxides represent an emerging class of cathode active materials. The representative compound ZnMnO3 has sufficient capacities for superlong cycles at elevated temperatures, owing to the suppression of the undesired spinel-rocksalt phase transition. To further enhance the electrochemical performance of ZnMnO3, hydrothermal treatment was applied to synthesize fine ZnMnO3 nanoparticles. By controlling the pH of precursor solutions, treatment temperature, and reaction duration, fine ZnMnO3 nanoparticles with a remarkably large surface area can be obtained. Our ZnMnO3, prepared using hydrothermal treatment, was able to deliver larger capacities compared with those obtained using the typical coprecipitation method. Furthermore, the hydrothermally treated ZnMnO3 allowed stable battery cycling even at 30 °C; this was achieved by combining the highly efficient, electrochemically stable electrolyte and the ZnMnO3 nanoparticles with the shortened diffusion path.

1. Introduction
Magnesium (Mg) metal rechargeable batteries (MRBs) are a promising candidate for next-generation batteries primarily owing to the remarkably high volumetric capacity of Mg metal (3837 mAh cm−3), which is approximately 10 times higher compared with a graphite anode.1 The cost-effectiveness and inherent stability of bulk Mg metal in an ambient atmosphere are also preferable for the mass production of large-scale energy storage devices based on MRBs. Important research on prototype MRBs was reported in 2000 where a certain Chevrel-phase compound (e.g., Mo6S8), Mg metal, and the tetrahydrofuran solutions of Mg organohaloaluminate salt (e.g., Mg(AlCl2BuEt)2) were used as a cathode, anode, and electrolyte, respectively.2 Since this pioneering work, several efforts have been made to develop novel cathode,3–12 anode,13–15 and electrolyte materials16–20 to realize practical MRBs. However, research into MRB materialization remains in a primitive stage because of several serious issues arising from the intrinsic chemical and electrochemical characteristics of Mg metal anodes and Mg2+ (Mg ions).
The fundamental electrochemical reactions in typical MRBs are the insertion and extraction of Mg2+ into/from cathode materials and the dissolution and deposition of Mg metal anodes. Because of the remarkably high surface charge density of Mg2+, the host materials (e.g., transition metal oxides) strongly bind to Mg2+, thereby leading to sluggish insertion and extraction reactions.21 To avoid such undesired situations, sulfide or selenide-based cathode active materials have been proposed.3–5,12,22–24 These compounds can effectively improve the reversibility and polarization of Mg2+ insertion and extraction processes. Using soft and heavy atoms, however, simultaneously leads to a lower operation potential, as well as insufficient deliverable capacities. Transition metal oxides should be the first option for achieving MRBs with high energy density beyond the presently available lithium-ion batteries. Among the variety of transition metal oxides, spinel compounds (represented as A(II)B(III)2O4) are regarded as promising oxide compounds in MRB applications.7,10,21,25
Upon discharging spinel oxide compounds, Mg2+ is inserted onto vacant 16c sites. This Mg2+ insertion is accompanied by the simultaneous push-out of A(II) ions occupying the 8a sites due to electrostatic repulsion between Mg2+ and A(II) ions, consequently leading to the structural changes from spinel-to-rocksalt phases. Inversely, Mg2+ is extracted from the 16c site, and A(II) ions will move back to the original 8a site with the recovery of spinel structures during the charging process. Electrochemical measurements combined with structural analysis techniques demonstrated the (quasi-)reversible structural change of certain spinel oxide compounds at elevated temperatures.10,25 However, this structural change will imperfectly proceed even at high temperatures, e.g., 150 °C; accordingly, the discharge capacities will diminish with cycling.25
One possible approach for improving cyclability is replacing Mg2+ at the 8a sites with Zn2+. Because Zn2+ prefers tetrahedral sites, the push-out of Zn2+ from the 8a to the 16c sites, accompanied by Mg2+ insertion, will be avoided, and the structural change between spinel and rocksalt phases should be suppressed. Compared with Mg-based counterparts, ZnCo2O4 and ZnFe2O4 indicated improved cyclability.26 The level of reversibility of these Zn-based spinel compounds, however, still appears to be insufficient for their practical use in MRBs.
Recently, another strategy for improving the cycling stability of spinel cathode materials was proposed, based on an introduction of defects in a spinel structure.10 ZnMnO3 is a representative defect spinel compound. In this structure, the 16c sites are entirely vacant, and part of the 16d site is not fully occupied. As Mg2+ can be inserted into partly unoccupied 16d sites, the irreversible transition between the spinel and rocksalt phases will be effectively suppressed. By combining Zn2+ as 8a site elements and introducing defects in a single spinel compound, i.e., ZnMnO3, the cycling stability was drastically improved; more than 150 cycles with negligible capacity decay were achieved.10
To further improve the electrochemical characteristics of ZnMnO3, a hydrothermal process was used in the present research to create fine nanoparticles comprising the desired compounds. Hydrothermal synthesis has several advantages, particularly in terms of controlling particle size and shape, as well as good industrial compatibility in mass production.27,28 Due to the large diffusion barrier of divalent Mg2+ in the oxide-based host lattice, the deliverable capacities of oxide cathodes strongly depend on the surface area of the primary particles;7 therefore, nanoparticles are particularly suitable for exploiting the full performance of active materials, particularly in MRBs. In this study, the effect of the experimental conditions in the hydrothermal treatment of ZnMnO3 was systematically investigated, concerning the pH of a precursor solution, reaction temperature, and reaction duration. The electrochemical characteristics of the resulting ZnMnO3 samples were examined to clarify the relationship between the physical characteristics of the active materials and battery performance. Using fine ZnMnO3 nanoparticles, we demonstrated the stable charge–discharge cycling of spinel-type oxide compounds at ambient temperature.
2. Experiment
2.1 Preparation of ZnMnO3
2.1.1 Hydrothermal process
The ZnMnO3 nanoparticles were synthesized using conventional hydrothermal processes. To highlight the benefits of hydrothermal processes on the electrochemical performance of ZnMnO3, the one synthesized by a typical coprecipitation method was also prepared.10 As the exchange between proton and metal ions is a primitive driving force of hydrothermal processes,29 proton activity dominated the overall reaction mechanisms and products. The reaction temperature and duration are also important factors for controlling the crystallinity, size, and chemical composition of the resulting compounds.28 To optimize the synthetic condition, the influence of three different parameters, i.e., proton activity, reaction temperature, and duration, on the purity and electrochemical characteristics of the resulting samples were systematically examined.
2.1.2 The effect of proton activity in the precursor solutions and reaction temperature
The ZnCl2 (0.523 mmol; 71.3 mg), MnCl2 (0.314 mmol; 39.3 mg), and oxalic acid (1.046 mmol; 94.2 mg) were dissolved in distilled water (10 mL); a 10 mol dm−3 sodium hydroxide solution was used to adjust the pH of the prepared precursor solutions to a pH of 10, 11, 12, and 13, respectively. Then, 0.2 mL of a 20 wt% tetraethylammonium hydroxide aqueous solution and KMnO4 (0.209 mmol; 33.0 mg) were added to each precursor solution. An autoclave (type TPR5; Taiatsu Glass Kokgyo Co.) was used to heat the precursor solutions at 110 °C, 130 °C, or 150 °C for 24 h with stirring. After the hydrothermal process, the prepared samples were collected using a freeze-dried method. The obtained samples were subjected to heat treatment at 200 °C for 3 h in an air atmosphere.
2.1.3 The effect of the reaction duration
For the impurity-less samples obtained according to certain conditions, the effect of the reaction duration on the physical characteristics of the obtained compounds was further investigated to optimize the synthetic condition. The same procedure as described above was adopted to synthesize ZnMnO3, except for the reaction duration. The duration was in the range of 1–72 h.
2.2 Physical characterization
The crystal structure of the prepared samples was investigated using powder X-ray diffraction (XRD) (Rigaku, SmartLab). All measurements were conducted using monochromatic CuKα radiation (λ = 1.54078 Å; 40 kV, 40 mA). The XRD measurements were conducted in the 2θ range from 10° to 70° with a step scan at 2° min−1 (step width, 0.02°). The collected diffraction data were compared to the database. The microstructure and morphology of the samples were observed using a scanning electron microscope (SEM; JEOL JSM-6490A). The elemental analysis of the same samples was conducted using energy-dispersive X-ray spectroscopy. The chemical compositions of the samples were determined via inductively coupled plasma spectrometry (Rigaku, CIROS-120). The specific surface areas were measured by applying the Brunauer–Emmett–Teller (BET) theory (MICROTRAC MRB, Belsorp II).
2.3 Electrochemical characterization
The electrochemical characteristics of the prepared samples were evaluated using three-/two-electrode setups. The different experimental setups were adopted based on the measurement temperature, i.e., an inorganic binary ionic liquid electrolyte with the three-electrode setup at 150 °C and a particular liquid electrolyte for the two-electrode setup at 30 °C.
For the three-electrode setup, ZnMnO3 as an active material, acetylene black (AB) as a conductive carbon, and polytetrafluoroethylene (PTFE) as a binder were mixed using mortar in the following weight ratio of active material : AB : PTFE = 60 : 30 : 10, followed by vacuum drying at 80 °C for 24 h. The dry composite was then compressed on Al mesh. The loading of active materials and the thickness of the composite electrodes were fixed at approximately 1 mg and 200 µm, respectively. The prepared composite and Mg alloy foil (AZ31; Mg : Al : Zn = 96 : 3 : 1 wt%) were served as a working and counter electrodes, respectively. The Ag+/Ag reference electrode was prepared according to the literature.30 The electrode potential of the Ag+/Ag reference electrode was calibrated using a typical Grignard reagent, i.e., approximately 2 mol dm−3 C2H5MgCl/tetrahydrofuran, to be −2.62 V vs. Ag+/Ag, which roughly corresponded to 0 V vs. Mg2+/Mg. The electrolyte was the binary mixture of Mg bis(trifluoromethanesulfonyl)imide (Mg(TFSI)2) and Cs(TFSI) with a 10 : 90 molar ratio.10,25 The discharge–charge tests were conducted using an automatic discharge and charge cycle unit (TOSCAT-3100, Toyo System Co.). The galvanostatic intermittent titration technique (GITT) was also adopted to evaluate the diffusion coefficient of Mg2+ in the composite electrode. The galvanostatic cycling and GITT measurements were started from the start of the discharge process. The cutoff voltages for the discharge and charge processes were prefixed at −1.1 V vs. Ag+/Ag (corresponding to approximately +1.52 V vs. Mg2+/Mg) and +0.6 V vs. Ag+/Ag (approximately +3.22 V vs. Mg2+/Mg), respectively. The discharge and charge currents for ZnMnO3 were fixed at 7.96 mA g−1. For GITT measurements, the discharge at each 7.96 mA g−1 for 15 min was followed by a 2 h rest at the open-circuit voltage. The diffusion coefficient was evaluated according to the voltage relaxation curves. Cell preparation and electrochemical measurements were performed in an argon-filled glove box with H2O < 0.1 ppm and O2 < 3 ppm.
For the two-electrode setup, the slurry composite of ZnMnO3 served as a working electrode; ZnMnO3 as an active material, AB as a conductive carbon, and PTFE as a binder were mixed using a mortar at a weight ratio of ZnMnO3 : AB : PTFE = 80 : 10 : 10. The resulting composite was pressed onto an Al mesh and then cut into a circular shape (ϕ = 14 mm). The composite cathode sheet was dried at 80 °C for at least 12 h under a vacuum. Then, 2032-type coin cells were assembled with the prepared composite as a cathode, and AZ31 (ϕ = 14 mm) was used as an anode. These electrodes were separated by two sheets of a homemade three-dimensionally ordered macroporous polyimide separator (ϕ = 18 mm). The 0.3 mol dm−3 Mg tetrakis(hexafluoro-iso-propyloxy)borate Mg[B(HFIP)4]2, which was dissolved in triglyme (G3), was served as an electrolyte.19 The discharge–charge cycling and GITT measurements were also conducted for the two-electrode setup to further evaluate the electrochemical characteristics of the ZnMnO3 at an ambient temperature. The same experimental conditions as those for the three-electrode setups were applied with the exceptions of the experimental temperature (30 °C) and the charge–discharge current densities (3.98 mA g−1).
3. Results and Discussions
3.1 Optimal synthetic conditions for ZnMnO3
In hydrothermal reactions, the ingredient components and ions in the reaction solutions dissolve and recrystallize repeatedly, resulting in progressive nucleation and crystal growth.29 During these processes, proton exchange occurs as a primitive stage. Therefore, synthetic conditions, such as the proton activity of the reaction solvent, hydrothermal treatment temperature, and treatment time, should have significant impacts on the composition and physical states of the final products. To reveal the optimal synthetic condition for the ZnMnO3 defect spinel, the above three experimental parameters were systematically investigated in this study.
For the fixed hydrothermal treatment temperature and reaction duration at 150 °C for 24 h, an impurity-less cubic ZnMnO3 was obtained from one specific precursor with pH = 13. As indicated by the XRD profiles (Fig. 1), tetragonal ZnMn2O4 impurities were generated in addition to the desired ZnMnO3 phase at a lower pH. The intensities of the peaks that were assignable to this impurity became prominent with decreasing pH values; conversely, the increasing proton activity of the precursors suggested that higher proton activity in the precursor solutions promoted undesired ZnMn2O4 generation during hydrothermal processes at 150 °C. Unfortunately, the synthetic reproducibility of the present condition was extremely poor for the ZnMnO3 preparation. This indicated that the composition of the products was dependent on the reaction batch.
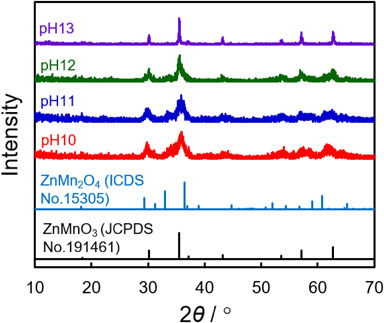
Tetragonal ZnMn2O4 is known to be thermodynamically more stable than the ZnMnO3 defect spinel. The intense diffraction peaks of ZnMn2O4 were observed for the sample prepared at 180 °C (Fig. S1). Accordingly, the relatively higher reaction temperature may have caused ZnMn2O4 generation. To suppress ZnMn2O4 generation, lower reaction temperatures were adopted during the hydrothermal treatments. The products of the hydrothermal treatment temperature at 130 °C were however found to be composed of only the ZnO phase (Fig. S1). The recent study proposed the hydrothermal reaction mechanism for ZnMnO3 generation from ZnO and MnO2 with a presence of KMnO4.31 The exchange of K+ from KMnO4 with proton will serve as a trigger for the structural decay and subsequent dissolution of Mn ions (from both MnO2 and KMnO4) into the reaction solution, resulting in a Zn–Mn complex formation, which will consequently lead to the nucleation of ZnMnO3. Based on this mechanism, the lower proton activity of the present reaction solution was potentially responsible for the prohibited ZnMnO3 conversion from the Zn and Mn precursors because the reaction rate of the ZnMnO3 nucleation was sensitive to proton activity. To control the reaction rate of the ZnMnO3 nucleation without inducing the formation of ZnMn2O4 and ZnO impurities, the pH values of the precursor solutions were modulated with a decrease in the reaction temperature, which resulted from the proton activity decreasing as the temperature diminished, owing to the principles of the equilibrium of H+ and OH− with H2O.
As summarized in Table 1, the impurity-less ZnMnO3 nanoparticles was obtained from the specific combinations of the hydrothermal temperature and the pH of the precursor solutions. At 130 °C, an impurity-less ZnMnO3 was obtained only from the pH 12 precursor, while the ZnMn2O4 impurities were simultaneously generated with ZnMnO3 formation from the lower pH precursors. With a decrease in the hydrothermal temperature, the precursor solutions required even lower pH values, e.g., pH 11, while the remaining pH conditions led to either ZnMn2O4 or ZnO impurity phase formation. Although the precise mechanisms (particularly for ZnMn2O4 formation at lower pH conditions) were still unclear, higher proton activity was found to have accelerated ion exchange reactions; this was followed by Zn–Mn complex nucleation at a relatively higher temperature, thereby inducing the formation of a thermodynamically quite stable ZnMn2O4 phase.
Table 1. Experimental conditions for the impurity-less ZnMnO
3.
Hydrothermal treatment temperature
/°C |
pH of the precursor solutions |
| 10 |
11 |
12 |
13 |
| 150 |
×a |
×a |
×a |
s.p. |
| 130 |
×a |
×a |
s.p. |
×b |
| 110 |
×a |
s.p. |
×b |
— |
s.p.: the conditions where the impurity-less ZnMnO3 are obtained; ×a: the products are composed of ZnMnO3 and ZnMn2O4 impurities; ×b: the reaction did not proceed, and ZnO was collected.
The physical properties of the ZnMnO3 obtained from the three different experimental conditions were characterized concerning the specific surface area and powder XRD profiles. The specific surface area increased with a decrease in the hydrothermal temperature and the pH of the precursor solutions (Table 2). The powder XRD profiles also reflected the difference in the crystallinity of the products as the profiles became broader with a decrease in the hydrothermal temperature and the pH of precursors (Fig. S2). These results strongly support the presence of ion exchange-driven nucleation and growth mechanisms. At a lower pH, owing to the relatively higher proton activity, the frequent ion exchange reactions may cause dominant nucleation rather than crystal growth, resulting in the formation of smaller crystallites. In contrast, the limited ion exchange reactions at lower levels of proton activity may induce crystal growth rather than nucleation, thereby leading to the formation of larger crystallites.
Table 2. BET surface area of the synthesized ZnMnO
3. Reaction duration was fixed at 24 h among the samples.
Hydrothermal treatment temperature
/°C |
Surface area/m2 g−1 |
| pH 10 |
pH 11 |
pH 12 |
pH 13 |
| 150 |
58.5 |
48.7 |
38.8 |
46.0 |
| 130 |
n.m. |
52.6 |
91.0 |
n.m. |
| 110 |
n.m. |
120.9 |
n.m. |
n.m. |
n.m.: not measured.
To further optimize the reaction condition for ZnMnO3, the effect of the reaction duration on the compositions and crystallinities of the resulting samples were investigated. According to the aforementioned results, the successful hydrothermal treatment temperature and the precursor pH conditions for the impurity-less ZnMnO3 were initially adopted. The reaction duration was found to strongly affect both the composition and crystallinity of the products. Overall, the insufficient reaction duration resulted in the incomplete conversion of MnO2 and ZnO into ZnMnO3. The corresponding XRD profiles observably indicate the presence of these reaction reagents in the samples obtained, which resulted from an insufficient reaction duration, i.e., less than 24 h for pH 12 at 130 °C and pH 11 at 110 °C (Fig. S3). Interestingly, the longer reaction duration did not affect the formation of impurities; however, an impact was observed on the crystallinities, implying that the activation energy barrier for ZnMn2O4 formation will be at approximately 130 °C for pH 12 and 110 °C for pH 11 precursors. The crystallinity observably improved with an increased reaction duration owing to the well-established sintering effect.
As described above, the specific surface area of the resulting samples was sensitive to the pH of the precursors, and a larger surface area could be achieved by decreasing the pH of these precursors. Generally, the surface area and grain size of the active cathode materials should be larger and smaller, respectively, to achieve favorable Mg2+ intercalation into the host lattice and subsequent diffusion within the lattice structure. Considering the thermodynamic stability of ZnMn2O4, its formation should be suppressed when controlling the reaction duration for the lower pH precursor solutions. Indeed, an impurity-less ZnMnO3 sample was successfully obtained by controlling the reaction duration even for the pH 10 precursors at a hydrothermal treatment temperature of 110 °C. The XRD profiles corroborated impurity-less ZnMnO3 formation from the pH 10 precursor that was hydrothermally treated at 110 °C for only 1 h (Fig. S4). The ZnMn2O4 formation was observed after increasing the reaction duration by several hours. Shortening the reaction duration with a decrease in the pH precursor was also effective for achieving a larger specific surface area. The surface area of ZnMnO3 obtained by the above specific condition reached 226.8 m2 g−1, which is twice as large as the other two conditions for ZnMnO3 (Table 3). The SEM images clearly show the formation of flower petal-like ultrathin crystal nano-assemblies with sub-nanometer dimensions for the former, while nontrivial granular crystals can be observed for the latter two samples (Fig. 2). A similar structure was also found for MgIn2S4 prepared using solvothermal treatment with a relatively shorter reaction duration.32 This coincidence strongly suggested that a minimized reaction duration would successfully suppress the agglomeration of crystalline nanosheets from becoming larger secondary particles.
Table 3. BET surface area of the synthesized ZnMnO
3.
| Experimental condition |
Code |
Surface area/
m2 g−1 |
pH of precursor/
treatment temperature/duration |
| pH 10/110 °C/1 h |
ZMO-10 |
226.8 |
| pH 11/110 °C/24 h |
ZMO-11 |
120.9 |
| pH 12/130 °C/24 h |
ZMO-12 |
91.0 |
Taking all of the experimental conditions into account, the impurity-less ZnMnO3 nanoparticles can be obtained under very limited conditions, as summarized in Table 3. The balance of proton activity, the solubility of starting reagents, and the thermodynamic stability of possible impurities will determine the composition and morphology of the final products. These different physical characteristics will affect the electrochemical characteristics of these products. The results of our electrochemical experiments are presented in the following section. The ZnMnO3 samples obtained through different experimental conditions are abbreviated hereafter as ZMO-10, ZMO-11, and ZMO-12 (Table 3).
3.2 Electrochemical characterization
To assess the electrochemical characteristics of a series of ZnMnO3, galvanostatic discharge–charge measurements were performed for ZMO-10, ZMO-11, and ZMO-12. Because of the poor compatibility of the inorganic ionic liquid-based electrolyte against Mg metal, a three-electrode setup with Ag+/Ag reference was used to control the working potential. Figure 3a shows the typical discharge–charge curves of the ZMO-10, ZMO-11, and ZMO-12 recorded at 150 °C. The initial discharge capacities were 156, 176, and 108 mAh g−1 for ZMO-10, ZMO-11, and ZMO-12, respectively (Fig. 3b). The capacities of the former two samples were remarkably larger than what was indicated in a previous report in which the same active material and electrolyte were used.10 This result strongly indicated an enhancement in the utilization ratio following micronization of the active materials as a result of the well-arranged hydrothermal treatments.
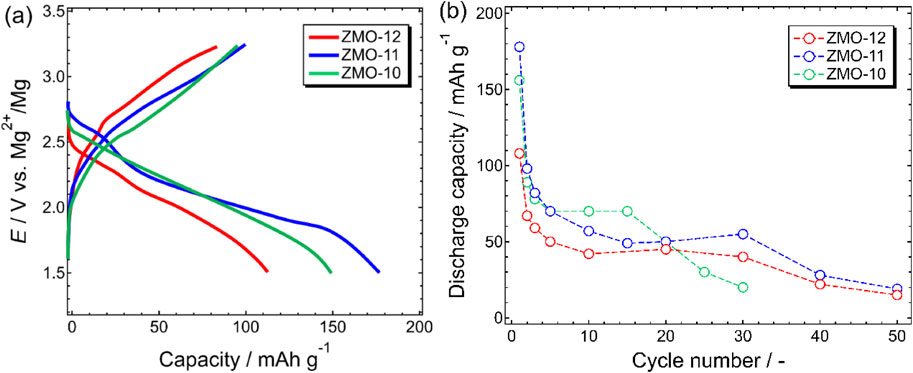
In stark contrast to the previous work,10 the deliverable discharge capacities of the subsequent cycles drastically diminished and finally reached approximately 20–30 mAh g−1 after 50 cycles (Fig. 3b). For the typical spinel oxide compounds in MRBs, the poor cycling stability is caused by the sluggish phase transition of the rocksalt (discharged) to spinel (charged) phases during Mg2+ extraction (charging) processes and the consequent uneven accumulation of the rocksalt phase in the samples.10,21 Such spinel-to-rocksalt phase transition is successfully suppressed upon combining the introduction of Zn to the 8a sites and defects in the O-sites for ZnMnO3. The XRD patterns of the ZMO cathode in different discharge–charge states demonstrated the suppression of undesired transitions from the defect spinel to the rocksalt phases (Fig. S5). That is, the capacity decay should not be caused by simple deterioration and/or phase transition of the cathode active material. To gain a more in-depth understanding of the possible causes of the poor cycling stability of the present sample, systematic studies concerning GITT measurements and EDX analysis were conducted.
The typical GITT profiles of the ZMO-12 recorded in the first and second cycles at 150 °C are shown in Fig. 4. The profiles of ZMO-10 and ZMO-11 are shown in Fig. S6. According to the GITT profiles, several important features were extracted. First, the profiles observably indicate a relatively large overpotential (>200 mV) during both the discharge and charge processes. The overpotential notably affected the deliverable capacities under the galvanostatic condition; the initial discharge capacity for the GITT measurement reached 170 mAh g−1, while that for the conventional galvanostatic discharge was 108 mAh g−1 (Fig. 3). Second, both the discharge and charge processes were found to comprise three stages. In the early stage, from the relaxed potential of 2.7–2.3 V vs. Mg2+/Mg, Mg2+ was inserted into or adsorbed onto the outermost surface of ZMO as the electrode potential rapidly dropped, even with a small degree of magnesiation. In the subsequent stage from 2.3 to 2.0 V vs. Mg2+/Mg, a stable, gentle slope was observed, indicating the single-phase reaction associated with the preferential Mg2+ insertion into cation deficiency sites.21 Following on, the spinel–rocksalt transition proceeded in the two-phase reaction as a final discharge stage. Similar profiles were observed for ZMO-10 and ZMO-11 while the discharge capacities were somewhat sample-dependent (Fig. S6). Third, undesired electrolyte decomposition occurred instead of the reverse rocksalt-spinel transition during charging. As evidenced by the gentle slope observed in the GITT profiles of the reverse charge scans, Mg2+ extraction proceeded well below approximately 2.8 V vs. Mg2+/Mg. The plateau observed at approximately 2.8 V vs. Mg2+/Mg, however, was attributed to the electrolyte decomposition; this was because almost the same charge profile was observed in the second charge, despite the considerably diminished discharge capacity of the corresponding process. The diffusion coefficients of Mg2+ ions ($D_{\text{Mg${^{2+}}$}}$), estimated from the relaxation behavior of the GITT curves, also implied that polarization possibly did not arise from the deterioration of the cathodes themselves. As summarized in Fig. 5, the $D_{\text{Mg${^{2+}}$}}$ values were almost in the same range, irrespective of processes and cycle number. The significant difference in the $D_{\text{Mg${^{2+}}$}}$ values at the initial discharge and charge stages may reflect the difference in the composition and crystal structures in the beginning of the discharge (ZnMnO3) and the charge (MgxZnMnO3) processes. For the capacity-dependent $D_{\text{Mg${^{2+}}$}}$, the values decrease as the progress of discharge and charge. During discharge, overpotential for Mg2+ intercalation gradually increases partly because of the phase-transition from spinel to rocksalt structures. For the early stage in the reverse charge process, Mg2+ can be de-intercalated from the crystalline lattice because binding from oxygen atoms should be relaxed by surrounding Mg2+ (and Zn, Mn) ions. With the progress of charge, which accompanies with increasing in the de-intercalated Mg2+ ions, Coulombic interactions between remaining Mg2+ and oxygen atoms would become strong, leading to large overpotential for de-intercalation. Consequently, the diffusion coefficients of Mg2+ during both discharge and charge decrease with the progress of each process. A similar profile of the diffusion coefficients has been observed for Zn2+ into/from ZnMn2O4.33 These observations indicate no or minor contributions on the part of the deterioration of the cathode active materials to polarization. Conversely, electrolyte decomposition after charging was supported by the EDX analyses. In the EDX mapping results for the pristine and cycled ZnMnO3 cathodes, the intense signals of S and Cs, which must be from the electrolyte, was detected for the cycled cathodes (Fig. S7). These results strongly corroborated severe electrolyte decomposition at the electrode–electrolyte interface, particularly during charging. The remarkably large surface area of the present active materials may facilitate interfacial electrolyte decomposition reactions while such large surface area of active materials will be effective in terms of enhancing electrochemical Mg2+ insertion/extraction reactions. It is noted here that the rocksalt phase made a minor contribution to the cyclability. Because of the large polarization, the cell voltage reached the predetermined cutoff voltage prior to the two-phase transition taking place. Although both single- and two-phase reactions will competitively occur, even in the early discharge stage,21 the XRD profile of the cathode in the later stage indicated the absence of the rocksalt phase (Fig. S5).

Regarding $D_{\text{Mg${^{2+}}$}}$ in the host materials, these values increased with an increase in the surface area of the samples (Fig. 6 and Table 3). For ZMO-10, which had the largest surface area among the studied samples, these $D_{\text{Mg${^{2+}}$}}$ values were approximately 5 times larger than for ZMO-12. According to the following equation,33,34
| \begin{equation*}
D_{\text{Mg${^{2+}}$}} = \frac{4}{\pi\tau}\left(\frac{mV_{\text{m}}}{MS}\right)^{2}\left(\frac{\Delta E_{\text{s}}}{\Delta E_{\tau}}\right)^{2}\quad \left(\tau \ll \frac{L^{2}}{D_{\text{GITT}}}\right)
\end{equation*}
|
where
τ is the duration time of the current pulse,
m is the mass of the active material,
M is the molecular weight and
Vm is its molar volume,
S is the total contacting area of electrode with electrolyte, Δ
Es is the difference in the open circuit voltage measured at the end of the relaxation period for two successive steps, Δ
Eτ is the difference in the voltage measured under the fixed current pulse applied, and
L is the thickness of the electrode, the diffusion coefficient should be independent of the surface area of the active material. Regarding the parameter
S, the “accurate” total contacting area of electrode with electrolyte should be calculated taking difference in the particle size into account. The simple geometric electrode area was however applied to the parameter
S in the present study because the accurate contacting area is extremely difficult to calculate for composite electrodes. Moreover, response of polarization against the current pulse, i.e. Δ
Eτ, was also influenced by the size of the active material even though Δ
Es should be identical at the same relaxation period independent of the samples. The observed difference in the diffusion coefficients among the studied three samples, ZMO-10, -11, and -12 may arise from these factors. It is noted here that the values of the present ZMO samples were in the range of 10
−9–10
−11 cm
−2 s
−1, which were comparable to the coefficients of Li
+ in LiFePO
4 at 150 °C.
35 Since LiFePO
4 is known to function as a representative cathode active material in lithium-ion batteries, even at ambient temperature, the ZMO nanoparticles are expected to be applicable as cathodes to MRBs operated at ambient temperature. For practical battery application, operation temperature should be moderate, e.g., ambient temperature, to enable adopting batteries for use in various devices. The recent drastic advancement of electrolyte materials offers the use of highly efficient and noncorrosive electrolytes. These electrolytes exhibit excellent compatibility against an Mg metal anode.
16–20 Owing to the absence of strong corrosive components, their anodic stability is high enough to be compatible with transition metal oxides.
7,8 In this study, the discharge–charge performance of ZnMnO
3 at ambient temperature was investigated using one such representative electrolyte, i.e., Mg[B(HFIP)
4]
2/G3.
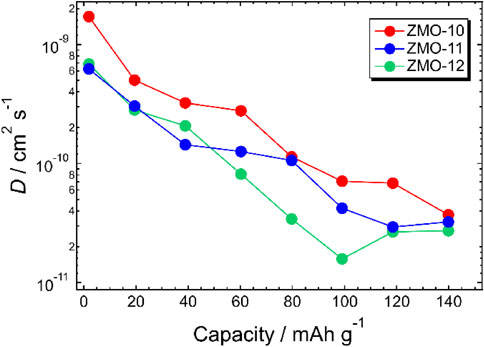
Figure 7 shows the discharge–charge curves of ZMO-12 recorded at 30 °C using a two-electrode setup in the 0.3 mol dm−3 Mg[B(HFIP)4]2/G3 electrolyte. Although the working voltage was lower for the two-electrode cells by approximately 1 V compared with the three-electrode counterparts (possibly because of the shift in the electrode potential of the Mg anode in the above-noted electrolyte), the two-electrode cells could be cycled even at ambient temperature and delivered sufficient capacities comparable to the control experiments with three-electrode cells performed at 150 °C (vide supra). The control experiment using the ZnMnO3 powder prepared by a coprecipitation method further emphasizes advantages of the hydrothermally treated ZnMnO3 as a cathode active material. The deliverable capacities and cycling stability of the latter was indeed significantly enhanced compared to the former one (Fig. S8). The capacity decay upon cycling also appeared to have been quite moderate for the cells cycled at 30 °C; this indicates the excellent electrochemical stability of the electrolyte during discharge–charge operations. We note that the discharge–charge performance of ZMO-12 was superior to that of MgMn2O4, which had a comparable surface area.7 The characteristic cation deficiency in the active material that would promote Mg2+ insertion/extraction may be responsible for the greater deliverable capacity and cycling stability of ZnMnO3.

The polarization during discharge–charge was, however, extremely large for the cells cycled at 30 °C. Such seriously large polarization was further emphasized by the GITT measurements. During both discharge and charge, a remarkably large voltage hysteresis of approximately 0.5–1.0 V was observed for the discharge, while this value was above 1.5 V for the charging process, suggesting the sluggish reactivity of the ZMO samples at ambient temperature, particularly for Mg2+ extraction (Fig. 8a). The $D_{\text{Mg${^{2+}}$}}$ estimated from the relaxation behavior of the GITT curves clearly supported the poor diffusivity of Mg2+ for these extraction processes (Fig. 8b). In the initial discharge/charge stages, the charge and discharge differences in $D_{\text{Mg${^{2+}}$}}$ appeared to have been moderate. With the increasing depth of each discharge and charge, however, this magnitude became much more prominent. As previously described, Mg2+ strongly bonded to the oxide-based lattice because of strong electrostatic Mg2+–O2− interactions; therefore, the diffusion of Mg2+ in such a lattice will be strongly hindered.21 The cation vacancies and small particle size jointly contributed to facilitating Mg2+ accommodation at the outermost surface. During the Mg2+ extraction (charge) processes, the Mg2+ located at the outermost surface could easily be extracted, while the Mg2+ diffusion inside would still be strongly hindered. This is partly why $D_{\text{Mg${^{2+}}$}}$ abruptly dropped in the moderate depth-of-charge phase.

The $D_{\text{Mg${^{2+}}$}}$ values of the representative sulfide-based cathode active materials of MRBs, i.e., Mo6S8 and MoS2, were estimated to be 6 × 10−12 and 4.4 × 10−12 cm−2 s−1,36,37 respectively, at ambient temperature. Since the $D_{\text{Mg${^{2+}}$}}$ for the present ZMO at 30 °C were quite large in the initial discharge–charge stages, ZMO can be regarded as a potential candidate in cathode active materials for MRBs, particularly those operated at ambient temperature. However, present battery performance remains insufficient for their practical application. To exploit the full potential of such promising oxide-based active materials for MRB applications, the physical characteristics of active materials themselves, as well as the composition of composite electrodes including conductive carbon and binder choices, cell fabrication, electrolytes, and Mg anodes and their assemblies, must be optimized.
4. Conclusion
The optimal experimental conditions for the hydrothermal treatment of impurity-less ZnMnO3 were systematically investigated concerning the pH of precursor solutions, treatment temperature, and reaction duration. The pH and treatment temperature of the solution were found to mutually affect the composition of the resulting samples, and high proton activity at a higher treatment temperature facilitated thermodynamically stable ZnMn2O4 impurity formation. Conversely, insufficient proton activity resulted in the incomplete conversion of the starting reagents into the desired compounds. The impurity-less ZnMnO3 nanoparticles with their remarkably large surface areas were successfully obtained through the appropriate modulation of proton activity and the treatment temperature, as well as a well-arranged reaction duration.
The ZnMnO3 prepared by hydrothermal treatment delivered larger capacities compared with those obtained by the typical coprecipitation method, which strongly depended on the surface area (particle size) of the samples. These capacities, however, rapidly diminished with cycling at higher operation temperatures. The GITT measurements (combined with EDX analysis) strongly suggested that it was not the deterioration of the cathode active materials themselves that caused large polarization during discharge–charge processes; rather, this was attributed to an increased interfacial resistance due to undesired electrolyte decomposition at the electrode–electrolyte interface, leading to a serious capacity decay with cycling. The diffusion coefficients of Mg2+ in ZnMnO3 were evaluated as being the same level as those of Li+ in LiFePO4 at 150 °C; this suggest the high potential of ZnMnO3 as an active cathode material for MRBs operated at ambient temperature. The battery performance at 30 °C with the highly efficient, electrochemically stable electrolyte was fascinating and achieved moderate discharge capacities and improved cycling stability. The remarkably large surface area and well-shortened diffusion path of ZnMnO3 (resulting from the formation of small nanoparticles because of the well-arranged hydrothermal treatment) jointly contributed to enhancing Mg2+ insertion/extraction activities. Although further studies and optimization in cell configuration are needed, the present study proposes hydrothermal treatment as a particularly effective approach for preparing active materials with a well-controlled composition and physical characteristics suitable for MRB applications.
Acknowledgment
The ZnMnO3 powder prepared by a coprecipitation method was kindly supplied by Dr. Tetsu Ichitsubo (Tohoku Univ.). This work was financially supported by the Advanced Low-Carbon Technology-Specially Promoted Research for Innovative Next Generation Batteries Program (ALCA-SPRING, Grant Number JPMJAL1301) and the NEXT Center of Innovation Program (COI-NEXT, Grant Number JPMJPF2016) of the Japan Science and Technology Agency (JST).
Data Availability Statement
CRediT Authorship Contribution Statement
Toshihiko Mandai: Data curation (Lead), Resources (Lead), Writing – original draft (Lead), Writing – review & editing (Lead)
Ayaka Kutsuma: Data curation (Lead), Formal analysis (Lead), Investigation (Lead)
Masashi Konya: Data curation (Lead), Formal analysis (Lead), Investigation (Lead)
Yukihiro Nakabayashi: Writing – review & editing (Lead)
Kiyoshi Kanamura: Conceptualization (Lead), Data curation (Lead), Funding acquisition (Lead), Project administration (Lead), Writing – review & editing (Lead)
Conflict of interests
There is no conflict of interests to declare.
Funding
Japan Science and Technology Agency (JST): JPMJAL1301 (Advanced Low Carbon Technology Research and Development Program - Specially Promoted Research for Innovative Next Generation Batteries (ALCA-SPRING))
Japan Science and Technology Agency (JST): JPMJPF2016 (NEXT Center of Innovation Program (COI-NEXT))
Footnotes
T. Mandai: ECSJ Active Member
K. Kanamura: ECSJ Fellow
References
- 1) Next Generation Batteries–Realization of High Energy Density Rechargeable Batteries (Ed., K. Kanamura), Springer Nature Singapore Pte Ltd., (2021).
- 2) D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich, and E. Levi, Nature, 407, 724 (2000).
- 3) Z. Zhao-Karger and M. Fichtner, Front. Chem., 6, 656 (2019).
- 4) L. Li, Y. Lu, Q. Zhang, S. Zhao, Z. Hu, and S.-L. Chou, Small, 17, 1902767 (2019).
- 5) Y. Shen, Y. Wang, Y. Miao, M. Yang, X. Zhao, and X. Shen, Adv. Mater., 32, 1905524 (2020).
- 6) C. Chen, J. Sun, L. Miao, Z. Yan, and J. Chen, Chem. Commun., 55, 14578 (2019).
- 7) K. Sone, Y. Hayashi, T. Mandai, S. Yagi, Y. Oaki, and H. Imai, J. Mater. Chem. A, 9, 6851 (2021).
- 8) K. Kajihara, D. Takahashi, H. Kobayashi, T. Mandai, H. Imai, and K. Kanamura, RSC Adv., 11, 19076 (2021).
- 9) N. Ishida, R. Nishigami, M. Matsui, T. Mandai, K. Kanamura, N. Kitamura, and Y. Idemoto, Electrochemistry, 89, 329 (2021).
- 10) K. Shimokawa, T. Atsumi, N. L. Okamoto, T. Kawaguchi, S. Imashuku, K. Wagatsuma, M. Nakayama, K. Kanamura, and T. Ichitsubo, Adv. Mater., 33, 2007539 (2021).
- 11) K. Qin, J. Huang, K. Holguina, and C. Luo, Energy Environ. Sci., 13, 3950 (2020).
- 12) X. Zhou, J. Tian, J. Hu, and C. Li, Adv. Mater., 30, 1704166 (2018).
- 13) J. Niu, Z. Zhang, and D. Aurbach, Adv. Energy Mater., 10, 2000697 (2020).
- 14) T. Mandai and H. Somekawa, Chem. Commun., 56, 12122 (2020).
- 15) H.-K. Tian, R. Jalem, M. Matsui, T. Mandai, H. Somekawa, and Y. Tateyama, J. Mater. Chem. A, 9, 15207 (2021).
- 16) O. Tutusaus, R. Mohtadi, T. S. Arthur, F. Mizuno, E. G. Nelson, and Y. V. Sevryugina, Angew. Chem., Int. Ed., 54, 7900 (2015).
- 17) J. T. Herb, C. A. Nist-Lund, and C. B. Arnold, ACS Energy Lett., 1, 1227 (2016).
- 18) Z. Zhao-Karger, M. E. G. Bardaji, O. Fuhr, and M. Fichtner, J. Mater. Chem. A, 5, 10815 (2017).
- 19) T. Mandai, ACS Appl. Mater. Interfaces, 12, 39135 (2020).
- 20) T. Mandai, Y. Youn, and Y. Tateyama, Mater. Adv., 2, 6283 (2021).
- 21) I. D. Johnson, B. J. Ingram, and J. Cabana, ACS Energy Lett., 6, 1892 (2021).
- 22) X. Sun, P. Bonnick, V. Duffort, M. Liu, Z. Rong, K. A. Persson, G. Ceder, and L. F. Nazar, Energy Environ. Sci., 9, 2273 (2016).
- 23) X. Sun, P. Bonnick, and L. F. Nazar, ACS Energy Lett., 1, 297 (2016).
- 24) Z. Zhang, Z. Cui, L. Qiao, J. Guan, H. Xu, X. Wang, P. Hu, H. Du, S. Li, X. Zhou, S. Dong, Z. Liu, G. Cui, and L. Chen, Adv. Energy Mater., 7, 1602055 (2017).
- 25) S. Okamoto, T. Ichitsubo, T. Kawaguchi, Y. Kumagai, F. Oba, S. Yagi, K. Shimokawa, N. Goto, T. Doi, and E. Matsubara, Adv. Sci., 2, 1500072 (2015).
- 26) K. Shimokawa, T. Atsumi, M. Harada, R. E. Ward, M. Nakayama, Y. Kumagai, F. Oba, N. L. Okamoto, K. Kanamura, and T. Ichitsubo, J. Mater. Chem. A, 7, 12225 (2019).
- 27) K. Kanamura, K. Dokko, and T. Kaizawa, J. Electrochem. Soc., 152, A391 (2005).
- 28) K. Dokko, S. Koizumi, H. Nakano, and K. Kanamura, J. Mater. Chem., 17, 4803 (2007).
- 29) Handbook of Hydrothermal Technology (Eds., K. Byrappa and M. Yoshimura), Noyes Publications, Park Ridge, NJ (2001).
- 30) T. Mandai, Y. Akita, S. Yagi, M. Egashira, H. Munakara, and K. Kanamura, J. Mater. Chem. A, 5, 3152 (2017).
- 31) F. Geng, A. Yuan, and L. Xu, Electrochim. Acta, 216, 376 (2016).
- 32) Y. Zhang, M. Konya, A. Kutsuma, S. Lim, T. Mandai, H. Munakata, and K. Kanamura, Small, 15, 1902236 (2019).
- 33) N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu, and J. Chen, J. Am. Chem. Soc., 138, 12894 (2016).
- 34) Z. Shen, L. Cao, C. D. Rahn, and C.-Y. Wang, J. Electrochem. Soc., 160, A1842 (2013).
- 35) R. Amin, J. Macier, P. Balaya, D. P. Chen, and C. T. Lin, Solid State Ionics, 179, 1683 (2008).
- 36) M. D. Levi, E. Lancry, H. Gizbar, Y. Gofer, E. Levi, and D. Aurbach, Electrochim. Acta, 49, 3201 (2004).
- 37) Y. Liang, H. D. Yoo, Y. Li, J. Shuai, H. A. Calderon, F. C. R. Hernandez, L. C. Grabow, and Y. Yao, Nano Lett., 15, 2194 (2015).



