Abstract
Catechols and their metal complexes are known to participate in electron-transfer reactions in diverse fields. However, most studies have been limited to dioxolene complexes in which the central metal and catechol moieties are directly linked via two adjacent oxygen atoms. Because catechol has oxygen atoms at adjacent positions the oxygen atoms can serve as coordination atoms. In this study, manganese(I) diimine(tricarbonyl) complexes with a free catechol unit are unprecedentedly synthesized to generate an oxidized form, o-quinone, on the complexes. Two types of monodentate ligands are used to control the electronic states of the complexes. Complexes containing the reduced (catechol) form are successfully isolated and characterized using spectroscopic and crystallographic analyses. The redox-responsive nature of the Mn complexes is confirmed by electrochemical analysis. The redox-induced interconversion between the catechol and o-quinone units is observed only in the complex, using electrochemical techniques. This study paves the way for the in situ electrochemical formation of redox-active, unstable organic compounds. Furthermore, the experimental methodologies described herein can establish the redox properties of the resulting species.

1. Introduction
Catechol (1,2-benzenediol), the disubstituted form of benzene, is a well-known redox-active organic compound. Catechol is interconverted to the corresponding o-benzoquinone via two-electron/two-proton transfers (Scheme 1), in a process called proton-coupled electron-transfer (PCET). PCET is a biologically important reaction and is involved in electron transport systems in photosynthesis and respiration.1 Metal complexes which comprise catechol as a ligand have been widely studied because of these specific properties. However, because catechol has oxygen atoms at adjacent positions that can be coordination atoms, almost all studies have been limited to dioxolene complexes, in which the central metal and catechol moieties (more precisely, deprotonated dianionic compounds) are directly linked via two adjacent oxygen atoms.2–6 By contrast, metal complexes with free catechol or o-benzoquinone units that are not directly attached to the metal center can retain their function as redox-active organic compounds. Thus, they provide characteristic functions such as visible-light responses and the precise control of electronic states in metal complexes.

To date, a variety of transition metal complexes containing dpq (1,10-phenanthroline-5,6-dione) as a ligand in which an o-benzoquinone unit is attached to the 1,10-phenanthroline (phen) framework (one of the typical α-diimine ligands) has been reported.7–10 Manganese, in particular, is expected to be used on an industrial scale in the future11 because it is inexpensive and more abundant than ruthenium or platinum.12 Previously, we reported the interconversion between quinone and catechol on a complex by various electrochemical experiments on a manganese(I) tricarbonyl complex containing the dpq ligand ([MnBr(CO)3(dpq)]).13 A reductive reaction system can be constructed from the quinone because dpq contains a stable quinone (oxidized) form. However, few studies have examined the oxidation reactions of α-diimine complexes containing a catechol (reduced) form.13,14
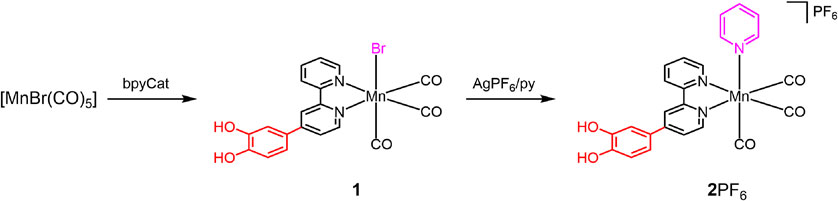
In this work, we focused on 4-(3,4-benzenediol)-2,2′-bipyridine (bpyCat), in which the stable catechol unit is introduced into the 2,2′-bipyridine (bpy) framework (which is a representative α-diimine similar to phen) as a ligand.8 bpyCat-containing manganese(I) complexes ([MnBr(CO)3(bpyCat)]: 1) were prepared and fully characterized; the characterization included structural analysis (Fig. 1). In addition, another complex with a monodentate ligand replaced with pyridine (py) from Br− [Mn(py)(CO)3(bpyCat)]+ (2+) was prepared to control the oxidation potential. Using these complexes, we performed molecular conversions from the catechol form to the corresponding quinone on manganese complexes based on the electrochemical oxidation reactions of the catechol unit. Therefore, we created new reactivities that are not expressed by organic compounds alone based on molecular designs that contain redox-active sites such as free catechols and quinones on metals.
2. Experimental
2.1 Materials and measurements
All chemicals were used without further purification, unless otherwise noted. All solvents used for the organic synthesis were anhydrous and used without purification. CH3CN for the electrochemical measurements was purified using a glass-contour solvent system (Laguna, CA, USA). The starting material ([MnBr(CO)5]) was purchased from Sigma-Aldrich (Tokyo, Japan). The precursors of bpyCat, 4-(3,4-dimethoxybenzene)-2,2′-bipyridine (bpyOMe), and the reference complex ([MnBr(CO)3(bpyOMe)]) were prepared according to previously reported procedures.7,15 The formation of these compounds was confirmed using MS, 1H NMR, and IR spectral data. All the manganese(I) complexes were handled and stored in the dark to minimize light exposure.
Microanalysis data were obtained using a Perkin Elmer 2400II series CHN analyzer (Yokohama, Japan). The 1H and 13C{1H} NMR spectra were recorded using a JEOL LMN-AL300 spectrometer (Tokyo, Japan) operating at 1H and 13C frequencies of 300 and 75.5 MHz, respectively. Both spectra were referenced to external (CH3)4Si via the residual protons of the solvent (1H) or the solvent itself (13C). Electrospray ionization mass spectrometry (ESI-MS) data were acquired using a micrOTOF instrument (Bruker Daltonics, Yokohama, Japan). Infrared (IR) spectra were obtained using the KBr pellet method on a JASCO FTIR 4100 spectrometer (Tokyo, Japan). Electronic absorption spectra were obtained in 1 cm quartz cuvettes using a JASCO V-560 spectrophotometer (Tokyo, Japan). Density functional theory (DFT) calculations for 1 and 2+ were performed using the quantum chemical program Gaussian 09W.16 The geometries of the complexes were fully optimized using a restricted DFT method employing the B3LYP function17,18 with a LanL2DZ basis set for the Mn and Br atoms,19 and a 6-31G(d) basis set for the lighter elements (H, C, N, and O).20,21 The solvent effect of CH3CN was evaluated using an implicit solvent model and a polarizable continuum model. Vibration analyses were performed at the same calculation level as that used for the geometry optimization.
2.2 Synthetic procedures
2.2.1 Synthesis of bpyCat
The compound was prepared using a modified literature method.15 A freshly prepared bpyOMe (115 mg, 390 µmol) was added to CH2Cl2 (20 mL). After the addition of 4 mL of a BBr3 solution (1 mol dm−3 in CH2Cl2), the mixture was stirred for 1 h at −40 °C. The reaction temperature was slowly increased to ambient temperature, after which the mixture was stirred for 12 h. Distilled water (30 mL) was added to the solution, and the solvent was evaporated to dryness by heating. Addition of CH3OH (10 mL) to the residue resulted in the formation of a yellow suspension of bpyCat. The product was collected via filtration, washed with CH3OH, and dried in vacuo. Yield: 76 mg (73 %). The formation of bpyCat was confirmed by 1H NMR spectroscopy.
2.2.2 Synthesis of fac-[MnBr(CO)3(bpyCat)] (1)
A Schlenk flask was charged with [MnBr(CO)5] (36 mg, 130 µmol) and bpyCat (35 mg, 130 µmol) and degassed with CH3OH (25 mL). The resulting mixture was stirred at 40 °C for 8 h. Subsequently, the solvent was evaporated under reduced pressure. After adding diethyl ether (30 mL) to the residue, a suspension of 1 was collected by filtration, washed with diethyl ether, and dried in vacuo. Yield: 43 mg (75 %). Anal. calcd for [MnBr(CO)3(bpyCat)]·H2O: C19H14N2O6BrMn: C, 45.54; H, 2.82; N, 5.59. Found: C, 45.56; H, 2.80; N, 5.28. IR (KBr): 2024, 1933, 1919 cm−1 (νCO). 1H NMR (DMSO-d6): δ 9.74 (s, 1H), 9.27 (s, 1H), 9.19 (d, J = 5.7 Hz, 1H), 9.06 (d, J = 5.7 Hz, 1H), 8.90 (d, J = 8.4 Hz, 1H), 8.79 (s, 1H), 8.24 (t, J = 7.2 Hz, 1H), 7.87 (dd, J = 6.0, 1.2 Hz, 1H), 7.72 (t, J = 6.6 Hz, 1H), 7.43–7.40 (m, 2H), 6.94 (d, J = 8.4 Hz, 1H). 13C{1H} NMR (DMSO-d6): δ 155.43, 153.36, 153.27, 150.01, 148.25, 145.99, 139.09, 126.73, 126.14, 123.54, 122.76, 119.40, 119.22, 116.10, 114.49.
2.2.3 Synthesis of fac-[Mn(py)(CO)3(bpyCat)]PF6 (2PF6)
Compound 1 (24 mg, 50 µmol) was dissolved in CH3CN (15 mL) under a N2 atmosphere. A solution of AgPF6 (12 mg, 50 µmol) in CH3CN (10 mL) was added, and the mixture was stirred at ambient temperature for 1 h. The resulting precipitate (AgBr) was filtered through a Celite column and the filtrate was removed using a rotary evaporator. CH3OH (20 mL) and 20 equiv. of pyridine (83 µL) were added, and the mixture was refluxed for 1 h. After cooling to ambient temperature, the solution was reduced to 2 mL using a rotary evaporator. The solution was allowed to stand overnight at −20 °C following the addition of diethyl ether. After filtering the insoluble matter, the filtrate was evaporated to dryness. The crude product was purified by trituration with diethyl ether several times to afford the yellow solid of 2PF6 with a yield of 29 mg (71 %). Anal. calcd for [Mn(py)(CO)3(bpyCat)]PF6·H2O·(CH3)2CO: C27H25N3O7PF6Mn: C, 46.10; H, 3.58; N, 5.97. Found: C, 46.18; H, 3.29; N, 5.67. ESI-MS (CH3CN): m/z 482 ([M]+). IR (KBr): 2038, 1934 cm−1 (νCO). 1H NMR (DMSO-d6): δ 9.85 (s, 1H), 9.43 (d, J = 4.5 Hz, 1H), 9.28 (d, J = 6.3 Hz, 1H), 9.23 (s, 1H), 8.87 (d, J = 7.8 Hz, 1H), 8.75 (s, 1H), 8.31 (d, J = 5.4 Hz, 1H), 8.04 (d, J = 7.3 Hz, 1H), 7.91–7.85 (m, 2H), 7.46–7.36 (m, 4H), 6.93 (d, J = 8.7 Hz, 1H). Attempts to obtain 13C{1H} NMR data for 2+ were unsuccessful because of its instability in solution.
2.3 Electrochemical methods
Electrochemical measurements were performed using an ALS/Chi model 660E electrochemical analyzer (Tokyo, Japan) with a solution of the complex in CH3CN (1 mmol dm−3) and n-Bu4NClO4 (0.1 mol dm−3) as the supporting electrolyte in a one-compartment cell consisting of a glassy carbon working electrode (ϕ = 1.6 mm), Pt wire counter electrode, and Ag|AgNO3 (0.01 mol dm−3 in CH3CN) reference electrode.22 Sufficient removal of O2 was confirmed by the absence of its reduction wave based on the background scans. Controlled-potential electrolysis was performed in two-compartment cells using a Pt mesh working electrode with a Pt spiral counter electrode in the second compartment separated by a glass frit. The number of electrons consumed during electrolysis (n) was determined by coulometric measurements. All potentials were reported in volts vs. the ferrocenium/ferrocene couple (Fc+/Fc) under argon at 25 °C. Fc+/Fc data were collected daily at the end of each experiment.
2.4 X-ray crystallographic analyses
Single crystals of bpyOMe were obtained via recrystallization from a dichloromethane/n-hexane mixture (1 : 1). Single crystals of bpyCat, 1, and [MnBr(CO)3(bpyOMe)] were obtained via vapor diffusion of n-hexane into acetone solutions of the compounds. Data for bpyOMe (at 293 K), bpyCat, 1, and [MnBr(CO)3(bpyOMe)] (at 153 K) were collected using a Rigaku Saturn CCD diffractometer (Tokyo, Japan) with graphite-monochromated Mo Kα radiation. All data were collected and processed using CrystalClear.23 The structures of all compounds were solved using direct methods and refined via full-matrix least squares on F2 using SIR9224 (bpyCat) or SHELXL25 (other compounds) in the CrystalStructure 4.3 software package.26 The crystallographic data are summarized in Tables S1 and S2 of the Supporting Information. CCDC-2286482, 2286483, 2286493, and 2286494 contain the supplementary crystallographic data for this paper.
3. Results and Discussion
3.1 Synthesis and spectroscopic properties
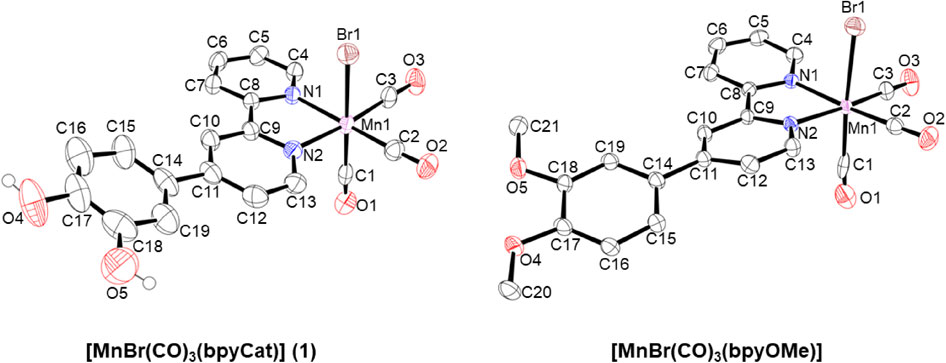
Although the synthesis of bpyCat, in which the 4-position of 2,2′-bipyridine is replaced by catechol, has been reported,15 the yield of bpyCat was improved in this study by optimizing several reaction conditions. As described later, the molecular structure of bpyCat was established using single-crystal X-ray structural analysis, which confirmed the predominance of the reduced form, even in air. By contrast, the dpc ligand (dpc = 5,6-dihydroxy-1,10-phenanthroline), is easily oxidized in air to afford dpq on the complex.13 The prepared bpyCat reacted with the precursor, [MnBr(CO)5], and produced 1 (Fig. 2), which contains one molecule of bpyCat. Furthermore, 2PF6, in which the bromido ligand was replaced with pyridine, was synthesized.
CO stretching vibrations (νCO) derived from coordinated carbonyls were observed in the IR spectra of 1 and 2PF6 (Table 1). The vibrational modes in Table 1 suggest that three carbonyl ligands coordinate in the facial position.27 The νCO peaks obtained for 2PF6 are higher energy region than those obtained for 1, because of the difference in the electronic states of the monodentate ligands (Br− or py).
Table 1. Spectroscopic data for
1 and
2+.
| |
1 |
2+ |
| νCOa/cm−1 |
2027 |
2038 |
| 1933 |
1934b |
| 1919 |
|
| λmaxc/nm (ε/103 mol−1 dm3 cm−1) |
292 (20) |
293 (26) |
| 326 (15) |
330 (15) |
| 421 (3.6) |
390 (7.5) |
| δ(OH)d/ppm |
9.75 |
9.87 |
| 9.26 |
9.24 |
a KBr method. b broad peak. c In CH3CN. d In DMSO-d6.
The absorption spectrum of 1 in CH3CN exhibits a broad absorption band due to the halogen-to-ligand charge transfer (XLCT) band, in addition to the metal-to-ligand charge transfer (MLCT) band (Fig. S1).7,28,29 However, the MLCT band is blue-shifted due to the substitution to pyridine in 2+, which is a π-acceptor ligand. The absorption spectra of 1 and 2+ did not change after being measured again after 1 and 2+ were stirred in air for 5 h, and it could be concluded that the catechol units in the complexes were stable even in solution.
The OH proton signals in the catechol unit were clearly observed as singlets in the 1H NMR measurements (δ 9.75 and 9.26 ppm for 1, δ 9.87 and 9.24 ppm for 2+) (Table 1). These signals disappeared completely upon the addition of D2O (Fig. S2).
3.2 Molecular structures of the ligands and complexes
X-ray structural analyses were performed on bpyCat and its precursor (bpyOMe). Additionally, the structures of 1 and the reference complex ([MnBr(CO)3(bpyOMe)]) were analyzed. The basic framework, 2,2′-bpy, was substituted at the 4-position in bpyOMe and bpyCat (Fig. 3). The C-OH bond lengths in bpyCat are similar to those of the corresponding moieties (C-OCH3) in bpyOMe, suggesting that bpyCat retains a catechol unit which contains C-O single bonds (Table 2).

Table 2. Selected bond lengths (Å) and angles (°) for the synthesized compounds.
| |
bpyCat |
bpyOMe |
1 |
[MnBr(CO)3(bpyOMe)] |
| Bond lengths |
C14-O1 |
1.378(3) |
C14-O1 |
1.359(4) |
C17-O4 |
1.379(7) |
C17-O4 |
1.355(4) |
| C15-O2 |
1.353(3) |
C15-O2 |
1.363(4) |
C18-O5 |
1.357(8) |
C18-O5 |
1.367(5) |
| C14-C15 |
1.390(3) |
C14-C15 |
1.398(5) |
Mn1-Br1 |
2.5459(7) |
Mn1-Br1 |
2.5085(10) |
| C8-C11 |
1.486(4) |
C8-C11 |
1.474(5) |
Mn1-N1 |
2.041(3) |
Mn1-N1 |
2.046(3) |
| |
|
C17-O1 |
1.406(5) |
Mn1-N2 |
2.027(3) |
Mn1-N2 |
2.056(3) |
| |
|
C18-O2 |
1.410(5) |
Mn1-C1 |
1.834(4) |
Mn1-C1 |
1.870(5) |
| |
|
|
|
Mn1-C2 |
1.809(4) |
Mn1-C2 |
1.802(4) |
| |
|
|
|
Mn1-C3 |
1.816(4) |
Mn1-C3 |
1.810(4) |
| |
|
|
|
C1-O1 |
1.070(5) |
C1-O1 |
1.007(6) |
| |
|
|
|
C2-O2 |
1.140(5) |
C2-O2 |
1.142(5) |
| |
|
|
|
C3-O3 |
1.139(5) |
C3-O3 |
1.141(5) |
| Bond angles |
|
|
C14-O1-C17 |
118.3(3) |
Mn1-C1-O1 |
177.0(3) |
Mn1-C1-O1 |
174.8(4) |
| |
|
C15-O2-C18 |
118.2(3) |
Mn1-C2-O2 |
176.3(3) |
Mn1-C2-O2 |
178.2(3) |
| |
|
|
|
Mn1-C3-O3 |
177.8(3) |
Mn1-C3-O3 |
177.4(4) |
Three CO ligands were confirmed to be facially coordinated to the central Mn atom in 1, as indicated by the IR data (Fig. 4). In addition, the bond parameters (Mn-C, Mn-N, Mn-Br, C-O lengths and Mn-C-O angles in Table 2) of 1 and the reference complex were similar to those reported for other fac-[MnBr(CO)3(N^N)]-type complexes (N^N = bidentate polypyridyl ligands).28–36 For the catechol unit in 1, the C-OH bond lengths (1.379(7) and 1.357(8) Å; Å = 10−10 m) were similar to those of a previously reported similar complex ([MnBr(CO)3(dpc)]; 1.362(2) and 1.367(2) Å),37–41 suggesting that the catechol form was retained in 1. This phenomenon was supported by the data (1.355(4) and 1.367(5) Å) obtained for the single-bonded methoxy groups (C-OCH3) in the reference complex.
3.3 Redox properties of the complexes and an attempt at accomplishing the electrochemical interconversion between catechol and quinone on the complex
The formation of the corresponding quinone form of bpyCat has not yet been reported to the best of our knowledge. In addition, all our attempts to synthesize the quinone forms chemically and electrochemically were unsuccessful. Therefore, the redox behavior of 1 and 2+, in which bpyCat was introduced into the complexes, was investigated in detail (Table 3).
Table 3. Electrochemical data for
1 and
2+ in CH
3CN (vs. Fc
+/Fc).
| |
1 |
2+ |
| Epa1/V |
0.69 (0.54) |
0.74 (0.61) |
| Epa2/V |
— (0.68) |
1.02 (1.02) |
| Epca/V |
0.05 (−0.04) |
−0.21 (−0.24) |
Data in parentheses represent redox potentials in the CH3CN/ethanol (7 : 1) solution.
a Subsequent reduction waves caused by oxidation of the catechol unit.
When cyclic voltammetry (CV) of 1 was performed in CH3CN, an irreversible oxidation wave was observed at 0.69 V (vs. Fc+/Fc) and a small reduction wave was observed at 0.05 V (Fig. 5a). By contrast, for 2+, which has the monodentate ligand replaced by py, two successive oxidation waves were observed at 0.74 V and 1.02 V, and a small reduction wave appeared near 0 V (Fig. 5b). Catechols and their derivatives generally exhibit two-step oxidation process in hydrophobic solutions. Additionally, catechols are sensitive to the acidity/basicity of the solution because they are typical Lewis basic aromatic compounds.42 Moreover, an irreversible oxidation wave due to MnII/I is observed at 0.69 V in the analogous complex, [MnBr(CO)3(bpy)].43 To clarify these oxidation waves, we performed CV measurements under a different set of conditions. Gutmann’s donor number (DN) serves as a measure of solvent basicity.44 When we added a more basic solvent, ethanol (DN = 20), into the acetonitrile solution (DN = 14.1) of 1 or 2+, only the oxidation waves assignable to the catechol unit were shifted to a negative potential (Table 3). From these results, the oxidation wave at 0.69 V in 1 can be considered to overlap the oxidation wave of catechol to quinone with that of MnII/I. In 2+, by contrast, the first wave (Epa1) can be assigned to the two-electron oxidation of catechol to quinone, and the subsequent wave (Epa2) is a MnII/I one-electron oxidation, which is supported by DFT calculations (Table S3). The voltammogram of 2+ can be interpreted as that of the oxidation from the catechol form to the corresponding quinone at 0.74 V; the resulting quinone-to-catechol reduction occurs near 0 V because the reduction wave appearing near 0 V is a subsequent wave of 0.74 V oxidation. This sequence of processes corresponds to “square scheme” in Scheme 2.45–47 The change in the potential for MnII/I is reasonably correlated with the electron-donating/accepting abilities of the monodentate ligand (Br−: π-donor, py: π-acceptor). Substitution of these ligands has an impact on the metal center and shifts the dπ orbital energies of the manganese(I). Thus, we confirmed that the difference in the monodentate ligands (Br− or py) has a marked effect on the oxidation potential of the metal center in particular.


CV measurements clarified that the oxidation potentials of the catechol unit and MnII/I were clearly separated in 2+. Therefore, we performed an electrochemical oxidation to the corresponding quinone via controlled-potential electrolysis. An exhaustive electrolysis of 2+ was performed at 0.90 V to selectively oxidize the catechol unit (Fig. 6a). When the CV was measured after two-electron oxidation (n = 1.9), the original oxidation wave (Epa1) due to the oxidation of the catechol unit disappeared, and the reduction wave accompanied by PCET of the quinone unit appeared at 0.05 V (Fig. 6b). The MnII/I potential was 1.04 V, which is similar to that prior to electrolysis (Epa2 = 1.02 V). The reductive electrolysis of the resulting quinone recovered the original catechol oxidation to 0.74 V again (Fig. 6c). The reversible structural changes between the two compounds based on electrolysis were also monitored by absorption spectroscopy (Fig. S3). These experiments suggest that the catechol unit electrochemically interconverts with the corresponding quinone on 2+. Additionally, the resulting quinone unit can exist stably in the test solution at 25 °C for more than 90 min. Therefore, in this study, we succeeded in electrochemically forming an unstable quinone on the complex and clarified its redox properties.

4. Conclusions
We demonstrated the electrochemical properties of Mn(I) complexes with a redox-active catechol unit attached to a bpy framework, which is a typical bidentate polypyridyl ligand. The oxidation potential of the Mn center was controlled by monodentate ligands with different electronic states; thus, the oxidation of the catechol unit could be performed selectively. As a result, the PCET-based conversion of catechol to quinone, which is difficult for organic compounds alone (bpyCat), was electrochemically observed in 2+. This work should lead to the in situ electrochemical formation of redox-active, unstable organic compounds and provide a series of experimental methodologies for determining the redox properties of the resulting species. Further studies are underway to investigate the interconversion of catechol and quinones into other complex systems.
Acknowledgments
We would like to thank Mr. Takatoshi Kanno at Fukushima University for technical assistance at an early stage of the project. The authors would like to thank Editage (www.editage.jp) for English language editing.
Data Availability Statement
CRediT Authorship Contribution Statement
Koki Chonan: Formal analysis (Lead), Investigation (Lead), Writing – original draft (Equal)
Tsugiko Takase: Data curation (Lead), Validation (Lead), Visualization (Lead)
Dai Oyama: Conceptualization (Lead), Funding acquisition (Lead), Methodology (Lead), Project administration (Lead), Supervision (Lead), Writing – original draft (Equal), Writing – review & editing (Lead)
Conflict of Interest
The authors declare no conflict of interest in the manuscript.
Funding
Fukushima University: foR-F
Footnotes
D. Oyama: ECSJ Active Member
References
- 1) D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty, and T. J. Meyer, Chem. Rev, 112, 4016 (2012).
- 2) C. G. Pierpont, Inorg. Chem., 50, 9766 (2011).
- 3) J. L. Boyer, J. Rochford, M.-K. Tsai, J. T. Muckerman, and E. Fujita, Coord. Chem. Rev., 254, 309 (2010).
- 4) T. Wada, T. Fujihara, M. Tomori, D. Ooyama, and K. Tanaka, Bull. Chem. Soc. Jpn., 77, 741 (2004).
- 5) K. Kobayashi, H. Ohtsu, T. Wada, T. Kato, and K. Tanaka, J. Am. Chem. Soc., 125, 6729 (2003).
- 6) C. G. Pierpont, Coord. Chem. Rev., 216–217, 99 (2001).
- 7) M. Stanbury, J.-D. Compain, M. Trejo, P. Smith, E. Goure, and S. Chardon-Noblat, Electrochim. Acta, 240, 288 (2017).
- 8) C. A. Goss and H. D. Abruna, Inorg. Chem., 24, 4263 (1985).
- 9) R. C. Conrad and J. V. Rund, Inorg. Chem., 11, 129 (1972).
- 10) W. J. Shi, Acta Crystallogr., Sect. E, 65, m653 (2009).
- 11) D. A. Valyaev, G. Lavigne, and N. Lugan, Coord. Chem. Rev., 308, 191 (2016).
- 12) H. Takeda, C. Cometto, O. Ishitani, and M. Robert, ACS Catal., 7, 70 (2017).
- 13) T. Kanno, T. Takase, and D. Oyama, Molecules, 25, 5921 (2020).
- 14) T. Fujihara, R. Okamura, T. Wada, and K. Tanaka, Dalton Trans., 3221 (2003).
- 15) S. Verma, P. Kar, A. Das, D. K. Palit, and H. N. Ghosh, J. Phys. Chem. C, 112, 2918 (2008).
- 16) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09W, revision D.01; Gaussian, Inc., Wallingford, CT, USA (2009).
- 17) C. Lee, W. Yang, and R. G. Parr, Phys. Rev. B, 37, 785 (1988).
- 18) A. D. Becke, J. Chem. Phys., 98, 5648 (1993).
- 19) W. R. Wadt and P. J. Hay, J. Chem. Phys., 82, 284 (1985).
- 20) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees, and J. A. Pople, J. Chem. Phys., 77, 3654 (1982).
- 21) W. J. Hehre, R. Ditchfield, and J. A. Pople, J. Chem. Phys., 56, 2257 (1972).
- 22) Safety note: Perchlorate salts are potentially explosive and should be handled with care.
- 23) Rigaku, CrystalClear, Rigaku Corporation, Tokyo, Japan (2015).
- 24) A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori, and M. Camalli, J. Appl. Crystallogr., 27, 435 (1994).
- 25) G. M. Sheldrick, Acta Crystallogr., Sect. C, 71, 3 (2015).
- 26) Rigaku, CrystalStructure, ver. 4.3: Crystal Structure Analysis Package, Rigaku Corporation, Tokyo, Japan (2019).
- 27) G. J. Stor, D. J. Stufkens, P. Vernooijs, E. J. Baerends, J. Fraanje, and K. Goubitz, Inorg. Chem., 34, 1588 (1995).
- 28) J. Jimenez, I. Chakraborty, and P. K. Mascharak, Eur. J. Inorg. Chem., 2015, 5021 (2015).
- 29) D. A. Kurtz, B. Dhakal, R. J. Hulme, G. S. Nichol, and G. A. N. Felton, Inorg. Chim. Acta, 427, 22 (2015).
- 30) M. Bourrez, F. Molton, S. Chardon-Noblat, and A. Deronzier, Angew. Chem., Int. Ed., 50, 9903 (2011).
- 31) W. C. Henke, C. J. Otolski, W. N. G. Moore, C. G. Elles, and J. D. Blakemore, Inorg. Chem., 59, 2178 (2020).
- 32) C. W. Machan, M. D. Sampson, S. A. Chabolla, T. Dang, and C. P. Kubiak, Organometallics, 33, 4550 (2014).
- 33) S. Lense, I. A. Guzei, J. Andersen, and K. C. Thao, Acta Crystallogr., Sect. E, 74, 731 (2018).
- 34) J. J. Walsh, C. L. Smith, G. Neri, G. F. S. Whitehead, C. M. Robertson, and A. J. Cowan, Faraday Discuss., 183, 147 (2015).
- 35) K. Wadayama, T. Takase, and D. Oyama, IUCrData, 4, x181792 (2019).
- 36) T. Kanno, T. Takase, and D. Oyama, Acta Crystallogr., Sect. E, 76, 1433 (2020).
- 37) C. J. Brown, Acta Crystallogr., 21, 170 (1966).
- 38) X. Y. Lin, S. J. Tang, and W. S. Wu, Acta Crystallogr., Sect. E, 65, o2367 (2009).
- 39) D. Oyama, M. Kido, R. Abe, and T. Takase, ChemistrySelect, 2, 2583 (2017).
- 40) E. C. Constable, G. Zhang, C. E. Housecroft, and M. Neuburger, Inorg. Chem. Commun., 13, 878 (2010).
- 41) B. Whittle, E. L. Horwood, L. H. Rees, S. R. Batten, J. C. Jeffery, and M. D. Ward, Polyhedron, 17, 373 (1998).
- 42) D. T. Sawyer, A. Sobkowiak, and J. L. Roberts, Jr., Electrochemistry for Chemists (2nd ed.), John Wiley & Sons, New York (1995).
- 43) S. Pordel and J. K. White, Inorg. Chim. Acta, 500, 119206 (2020).
- 44) V. Gutmann, The Donor-Acceptor Approach to Molecular Interactions, Plenum Press, New York (1978).
- 45) M. D. Stallings, M. M. Morrison, and D. T. Sawyer, Inorg. Chem., 20, 2655 (1981).
- 46) A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications (2nd ed.), John Wiley & Sons, New York (2001).
- 47) T. Kojima, Bull. Jpn. Soc. Coord. Chem., 74, 9 (2019). [in Japanese]