Abstract
Highly concentrated Li salt/aprotic solvent solutions are promising electrolytes for next-generation batteries. Understanding the Li+ ion transport process is crucial for designing novel battery electrolytes. In this study, we systematically investigated the phase behavior, solvate structures, and Li+ transport properties of binary mixtures comprising lithium bis(trifluoromethanesulfonyl)amide (LiTFSA) and various sulfones, such as sulfolane (SL), 3-methyl sulfolane (MSL), dimethyl sulfone (DMS), ethyl methyl sulfone (EMS), and ethyl isopropyl sulfone (EiPS). Except for the MSL system, the [LiTFSA]/[sulfone] = 1/2 mixtures remained in a liquid state at room temperature, thus enabling a systematic comparison of the Li+ transport properties in the highly concentrated electrolytes. In highly concentrated liquid electrolytes, Li+ ions diffuse by exchanging ligands (sulfone and TFSA). Li+ ions diffuse faster than TFSA in all electrolytes except the EiPS-based electrolyte at a composition of [LiTFSA]/[sulfone] = 1/2, resulting in high Li+ transference numbers. SL-based electrolytes show higher ionic conductivity and Li+ transference numbers than other sulfone-based electrolytes. Consequently, sulfone solvents with compact molecular sizes and low energy barriers of conformational change are favorable for enhancing the Li+ ion transport in the electrolytes.

1. Introduction
Mixtures of Li salts and aprotic solvents are used as electrolyte solutions in Li-ion batteries, and carbonate esters are widely used as aprotic solvents.1,2 Electrolyte solvents must have a wide electrochemical stability window and the ability to dissolve and dissociate high-concentration Li salts. Carbonate solvents exhibit high oxidation stability, and ethylene carbonate (EC) forms a stable passivation film on negative graphite electrode surfaces, preventing the subsequent reductive decomposition of the solvent and anions.3 Mixed solvents containing both cyclic carbonates, such as EC, and linear carbonates, such as ethyl methyl carbonate (EMC), are often used as electrolyte solvents. EC has a high polarity (relative permittivity εr ∼ 90) and can dissolve and dissociate Li salts, while linear carbonates exhibit low viscosity due to their low polarity (εr ∼ 3).4 As the conductivity of an electrolyte is determined by the number of ion carriers and their mobility, combining EC, EMC, and an Li salt in an appropriate ratio facilitates high ionic conductivity.5 Owing to these advantages, carbonate-based electrolyte solutions containing ∼1 mol dm−3 (M) Li salts have been commonly used in practical Li-ion batteries. Ether solvents exhibit good reductive stability; however, their oxidative stability is generally poor. Moreover, although nitrile and sulfone solvents possess good oxidative stability, they do not form passivation layers on graphite anodes in Li-ion batteries and hence are reductively decomposed. Therefore, these solvents have not been used as electrolytes in Li-ion batteries.
Recently, highly concentrated electrolytes containing solvents other than carbonates have attracted immense attention as promising electrolytes for next-generation batteries.6–14 The oxidative stability of the electrolytes improves by increasing the Li salt concentration.15 Moreover, an anion-derived passivation layer forms on the graphite anode in Li-ion batteries when Li salts with amide-type anions (LiN(SO2F)2 and LiN(SO2CF3)2) are used.16,17 This anion-derived passivation layer effectively suppresses the subsequent decomposition of the electrolyte. Consequently, highly concentrated electrolytes possess wider electrochemical stability windows than low-concentration electrolytes. Furthermore, Li-ion hopping conduction occurs in highly concentrated electrolytes that contain specific solvents and Li salts.7,18 In these highly concentrated electrolytes, the solvent molecules and anions crosslink Li+ ions to form Li+–solvent–Li+ and Li+–anion–Li+ network structures. Li+ ions dynamically exchange ligands (solvent molecules and anions) in these network structures and move faster than the ligands. To date, Li-ion hopping conduction has been observed in highly concentrated electrolytes that contain the following solvents: sulfolane (SL),18–23 dinitriles,24,25 keto esters,26 and γ-butyrolactone (GBL).27,28 Li-ion hopping conduction in electrolytes results in Li+ ion transference numbers (t+ > 0.6) that are higher than those in conventional carbonate-based electrolyte solutions (t+ < 0.3). The high Li+ ion transference number effectively suppresses the concentration overpotential in Li-ion batteries during high-rate charging and discharging.29,30
We recently reported the effects of the molecular structure of sulfones on Li+ ion transport properties in electrolyte solutions containing LiBF4/sulfone20 and LiN(SO2F)2/sulfone.21 In the present study, we investigated the physicochemical properties of binary mixtures composed of LiN(SO2CF3)2 (LiTFSA) and sulfone. Highly concentrated LiTFSA/SL electrolytes are promising electrolytes for Li–S batteries.31,32 Li–S batteries contain Li metal anodes and composite cathodes composed of molecular sulfur (S8), a carbon conductive agent, and a polymer binder. In these batteries, Li dissolution and the reduction of S8 occur at the anode and cathode, respectively, during discharging, and the reverse reactions occur during charging.33 In the case of Li–S batteries, lithium polysulfides (LiPSs) are formed as reaction intermediates at the S8 composite cathode during the discharging and charging processes. However, LiPSs dissolve into the conventional electrolytes containing 1 M Li salt, which lowers the Coulombic efficiency (CE) of the charge-discharge process and shortens the life of Li–S batteries.33–35 Highly concentrated LiTFSA/SL electrolytes can suppress the solubility of LiPSs and improve CE. In addition, Li-ion hopping conduction in electrolytes can suppress the concentration overpotential in Li–S batteries. However, LiBF4 and LiN(SO2F)2 react irreversibly with LiPSs, while LiTFSA is stable against LiPSs; therefore, LiBF4 and LiN(SO2F)2 are unsuitable electrolyte salts for Li–S batteries containing S8-based cathodes.33,36,37 In this paper, we report the phase behaviors and Li+ transport properties of binary mixtures composed of LiTFSA and various sulfone solvents, such as SL, 3-methyl sulfolane (MSL), dimethyl sulfone (DMS), ethyl methyl sulfone (EMS), and ethyl isopropyl sulfone (EiPS).
2. Experimental
Purified SL and EMS were purchased from Kishida Chemical and were used as received. DMS and EiPS were purchased from Tokyo Chemical Industry Co. EiPS was dried using molecular sieves (4A, Wako Chemical) before use. LiTFSA was supplied by Solvay. The electrolytes were prepared by simply mixing the Li salts and solvents in an Ar-filled glovebox (VAC, [H2O] < 1 ppm).
The thermal properties of the LiTFSA/sulfone mixtures were evaluated by differential scanning calorimetry (DSC) using the DSC6220 and DSC7020 instruments (Hitachi High-Tech Science). The DSC samples were sealed in aluminum pans in an Ar-filled glove box. The samples were first heated to an appropriate temperature to remove the thermal hysteresis, cooled to −150 °C, and finally heated to 60–120 °C at a rate of 5 °C min−1. Thermograms were recorded during the final heating process. The liquidus line temperature (Tliq) and glass transition temperature (Tg) were estimated from the maxima of the endothermic peaks and onset of changes in the heat capacity values, respectively.
X-ray crystallography was performed with an XtaLab P2000 diffractometer using monochromatic MoKα radiation (λ = 0.71073 Å). Single crystals of [LiTFSA]/[MSL] = 1/1 and [LiTFSA]/[EiPS] = 1/1 solvates were grown in [LiTFSA]/[MSL] = 1/2 and [LiTFSA]/[EiPS] = 1/1.5, respectively. The crystals were coated with vacuum grease to prevent contact with air and were mounted on a glass pin. Diffraction was measured at −50 °C under a steady flow of dry N2 gas. An empirical absorption correction was applied to the data via multi-scan averaging of the symmetry equivalent data using spherical harmonics implemented in the SCALES3 ABSPACK scaling algorithm (CrysAlisPro 1.171.39.46e, Rigaku Oxford Diffraction, 2018). Crystallographic structures were solved by the direct method using SHELXT. All nonhydrogen atoms were refined anisotropically via the full-matrix least-squares method using SHELXL-2018/3.38,39 All hydrogen atoms were placed at geometrically ideal positions and refined using an appropriate riding model.
A Raman spectrometer equipped with a 785-nm laser (NRS4100, JASCO) was used to record the Raman spectra of the electrolytes, which was calibrated using a polypropylene standard. Electrolyte samples were sealed in a capillary glass tube, and their temperature was maintained at 30 °C using a Peltier microscope stage (TS62, Instec) with a temperature controller (mK1000, Instec). After baseline correction, the integrated intensity of the bands derived from the solvent molecules around 1450 cm−1 (assigned to the CH2 bending/scissoring mode)40 was normalized.
The ionic conductivities (σ) of the electrolyte solutions were determined using electrochemical impedance spectroscopy with an impedance analyzer (VMP3, Biologic) in the frequency range of 500 kHz to 1 Hz with an amplitude of 10 mV root mean square (rms). A cell equipped with two platinized platinum electrodes (CG-511B, TOA Electronics) was used to measure the conductivity. The cell constant was determined using a KCl aqueous solution (0.01 M) at 25 °C. The cell was placed in a temperature-controlled chamber (SU-241, Espec) for at least 1 h prior to the measurements. The densities and viscosities of the electrolyte solutions were measured using a Stabinger viscometer (SVM 3000, Anton Paar).
Pulsed-field gradient nuclear magnetic resonance (PFG-NMR) spectra were obtained using a JEOL ECX-400 NMR spectrometer with a 9.4-T narrow-bore superconducting magnet equipped with a pulsed-field gradient probe and current amplifier. The electrolyte solution was injected to a height of approximately 5 mm into the NMR tube (BMS-005J Shigemi) to exclude the effects of convection, and the diffusivities of different components of the solution (solvent (1H, 399.7 MHz), anions (19F, 376.1 MHz), and Li+ ions (7Li, 155.3 MHz)) were evaluated at 30 °C. The detailed experimental procedure for PFG-NMR has been reported elsewhere.41,42
The Li+ transference number (tLi+abc) in the electrolyte solution under anion-blocking conditions was evaluated using a 2032-type coin cell with a symmetric configuration of [Li | electrolyte/separator | Li]. The cell assembly was carried out in an Ar-filled glove box. The Li-metal foil was purchased from Honjo Metal Co. and cut to a diameter of 16 mm. A glass-fiber filter with 200-µm thickness (GA-55, Advantec) was cut into 17 mm diameter pieces and used as a separator. Two Li-metal electrodes and a glass fiber filter impregnated with an electrolyte solution were encapsulated in a coin cell. Electrochemical measurements were conducted using an electrochemical analyzer (VMP3, Biologic), and the temperature of the cell was controlled using a thermostat chamber (SU-241). A constant voltage (ΔV = 10 mV) was applied to the Li/Li symmetric cell to record the chronoamperogram. AC impedance measurements were carried out for the same cell with an amplitude of 10 mV rms to evaluate the bulk electrolyte resistance (Rb) and the interfacial resistance at the Li metal electrodes. AC impedance measurements were conducted just before and after chronoamperometry. tLi+abc was calculated using the following equation:24,30
| \begin{equation}
t_{\text{Li+}}{}^{\text{abc}} = \frac{I_{\text{ss}}(\Delta V - I_{\Omega}R_{\text{i,0}})}{I_{\Omega}(\Delta V - I_{\text{ss}}R_{\text{i,ss}})}
\end{equation}
| (1) |
where
Iss is the steady-state current observed in the chronoamperogram, and
Ri,0 and
Ri,ss are the initial and steady-state interfacial resistances at the Li metal electrode, respectively.
IΩ is the ohmic current calculated from
Rb and the potential drop across the electrolyte at the steady state,
IΩ = Δ
V/(
Rb +
Ri, 0).
3. Results and Discussion
3.1 Phase behavior of LiTFSA/sulfone mixtures
Figure 1 shows the liquidus line temperature (Tliq) and glass transition temperatures (Tg) of the LiTFSA/sulfone binary mixtures as functions of the mole fraction of LiTFSA (XLiTFSA). The DSC curves of the mixtures are shown in Fig. S1 (Supporting Information (SI)). The melting point of pure DMS is 110 °C (Fig. 1a). The Tliq of LiTFSA/DMS decreased gradually with the increasing mole fraction of LiTFSA up to XLiTFSA = 0.29 (at a molar ratio of [LiTFSA]/[DMS] = 1/2.5). In addition, a solidus line temperature (Tsol) at approximately 20 °C and glass transition (Tg < −50 °C) were also observed. The Tliq of the [LiTFSA]/[DMS] = 1/2 mixture was 24.4 °C, which was slightly higher than Tsol, and a certain crystalline solvate may have been formed at this composition. Tliq was not observed for the [LiTFSA]/[DMS] > 1/2 mixtures, and the mixture became a glass-forming liquid. This observation is consistent with a previous report.43

The phase diagrams of the LiTFSA/EMS and LiTFSA/EiPS systems are shown in Figs. 1b and 1c, respectively. The liquidus line temperatures of these mixtures were relatively lower than those of the DMS-based system. The Tliq and Tg of the systems in the composition range of 0 ≤ [LiTFSA]/[sulfone] < 1 can be placed in the order: EiPS < EMS < DMS. These mixtures become glass-forming liquids over the wide composition ranges of [LiTFSA]/[EMS] > 1/5 and 1/10 < [LiTFSA]/[EiPS] < 1/1. The asymmetric structures of EMS and EiPS are favorable for lowering the liquidus temperature of the mixtures. In addition, both EMS and EiPS exhibit a higher freedom of conformation than DMS, which may have contributed to the lowering of Tliq. Notably, the [LiTFSA]/[EiPS] = 1/1 mixture exhibited a Tliq of 52.1 °C, indicating that a stable crystalline solvate was formed at this composition. Single crystals of [LiTFSA]/[EiPS] = 1/1 were grown in a mixture of [LiTFSA]/[EiPS] = 1/1.5 (vide infra).
The phase behavior of the LiTFSA/SL mixture has been reported previously.19 LiTFSA and SL form a crystalline solvate at the composition of [LiTFSA]/[SL] = 1/1. The melting points of pure SL and the [LiTFSA]/[SL] = 1/1 solvate are 27.5 and 52.9 °C, respectively. However, mixtures in the composition range of 0 < [LiTFSA]/[SL] ≤ 1/2 remain in a liquid state at room temperature. Figure 1d shows the Tliq and Tg values of the LiTFSA/MSL mixtures. The MSL-based mixtures melt at lower temperatures than the SL-based mixtures. The MSL-based mixtures did not exhibit Tliq in the composition range of [LiTFSA]/[MSL] > 1/10. It is likely that the crystallization was inhibited or slowed because MSL had an asymmetric structure and was a racemic mixture of two diastereoisomers. Although the liquidus line temperatures of the [LiTFSA]/[MSL] ≥ 1/2 mixtures were not observed in the DSC curves, these mixtures solidified after long-term storage at room temperature, and a single crystal of [LiTFSA]/[MSL] = 1/1 could be obtained from the [LiTFSA]/[MSL] = 1/2 mixture (vide infra).
X-ray crystallography was performed using the single-crystal sulfone solvates of LiTFSA. The crystal structures of [LiTFSA]/[EiPS] = 1/1 and [LiTFSA]/[MSL] = 1/1 are shown in Fig. 2, and their detailed crystallographic data are provided in Table S1. In the crystal structure of [LiTFSA]/[EiPS] = 1/1 (Fig. 2a), the coordination number of Li+ is 5. Each EiPS is coordinated to two different Li+ ions, and each TFSA− is also coordinated to two Li+ ions. In the crystal structure of [LiTFSA]/[MSL] = 1/1 (Fig. 2b), the coordination number of Li+ is 4. Each MSL molecule is coordinated to two different Li+ ions, which is similar to the case of [LiTFSA]/[EiPS] = 1/1. Each TFSA− anion is coordinated to two different Li+ ions via two -SO2− groups. Although the crystal structures of [LiTFSA]/[EiPS] = 1/1 and [LiTFSA]/[MSL] = 1/1 are different, the bridging structures of Li+–sulfone–Li+ and Li+–TFSA–Li+ are commonly observed. Network structures of Li+–sulfone–Li+ and Li+–anion–Li+ have also been reported for other sulfone solvates of Li salts.7,18,20,21

Raman spectroscopy was performed to assess the ionic association of LiTFSA in various sulfone-based solutions at 30 °C. As [LiTFSA]/[MSL] = 1/2 is solid at room temperature, the spectrum for this composition was recorded at 60 °C. In addition, LiTFSA/DMS mixtures containing low Li salt concentrations were solids at room temperature; thus, the spectra of the concentrated solutions of the LiTFSA/DMS mixtures with molar ratios of 1/2 or greater were collected. The peak position of the S-N-S symmetric stretching vibration is sensitive to the ionic interactions between TFSA− and the alkali metal cations.44–48 Figure 3 shows the concentration dependence of the Raman spectra of LiTFSA/sulfone solutions. The LiTFSA/sulfone solutions with low Li salt concentrations ([LiTFSA]/[sulfone] = 1/10) exhibited an almost constant wavenumber of 740.5 cm−1, irrespective of the sulfone structure. According to the literature,49 free TFSA and/or TFSA involved in the solvent-separated ion-pairs (SSIPs) in electrolyte solutions show an S-N-S vibration peak at 738–742 cm−1. Therefore, in low-concentration electrolytes with the [LiTFSA]/[sulfone] = 1/10 composition, the majority of TFSA anions are assumed to be not bound to Li+. In electrolyte solutions, solvents and anions (Lewis bases) competitively interact with Li+ ions (Lewis acids). The coordination number of Li+ in organic solvents is 4–5,44,50,51 which is consistent with the coordination number observed in the crystal structures shown in Fig. 2. The Gutman donor number (DN) is a good measure of Lewis basicity. The DNs of sulfones (DN ≥ 13)20 are higher than those of TFSA (DN ∼ 8);52 therefore, Li+ preferably coordinates with sulfone molecules when excess sulfone molecules are present in the mixtures. Thus, the interactions between the anions and Li+ are rather weak in solutions of [LiTFSA]/[sulfone] = 1/10 that contain excess amounts of sulfones. The S-N-S vibration peak shifts to higher wavenumbers when TFSA anions are coordinated to Li+.53 When the LiTFSA concentration increases, the distance between the cations and anions in the liquid decreases, leading to an enhanced attractive interaction between Li+ and TFSA−. As expected, the peak shifts to a higher wavenumber with increasing Li concentration ([LiTFSA]/[sulfone] > 1/6). The S-N-S vibration in all [LiTFSA]/[sulfone] = 1/2 mixtures exhibited similar Raman shifts in the range of 746–748 cm−1 (vertical lines in Fig. 3). In the [LiTFSA]/[sulfone] = 1/2 mixtures, the number of sulfone molecules is insufficient to fulfill the coordination number of Li+; therefore, the TFSA anions are also coordinated to the Li+ ions, resulting in the formation of contact ion pairs (CIPs) and/or ionic aggregates (AGGs), in which a single TFSA anion is coordinated to multiple Li+ ions.44,53 Further increasing the LiTFSA concentration to [LiTSFA]/[sulfone] = 1/1 causes a larger shift in the peak (approximately 750 cm−1), indicating that the number of AGGs has increased in the mixtures.49

The ionic conductivities and viscosities of the sulfone-based electrolytes are shown in Figs. 4a and 4b, respectively. The data for the SL-based electrolytes were obtained from a previous report.19 The ionic conductivity of the electrolyte increases with increasing XLiTFSA up to approximately 0.1 because of the increase in the number of charge carriers in the liquid. The viscosity of each electrolyte increases monotonically with increasing salt concentration. As shown in Fig. 4c, the diffusivity of Li+ (DLi) monotonically decreases with increasing salt concentration because of the increase in viscosity, resulting in a decrease in the mobility of ions. Although the number of charge carriers increases with increasing XLiTFSA, the decrease in the mobility of ions significantly affects the ionic conductivity of the solution of XLiTFSA > 0.1. This results in the volcano-type behavior of the conductivity-concentration relationship.
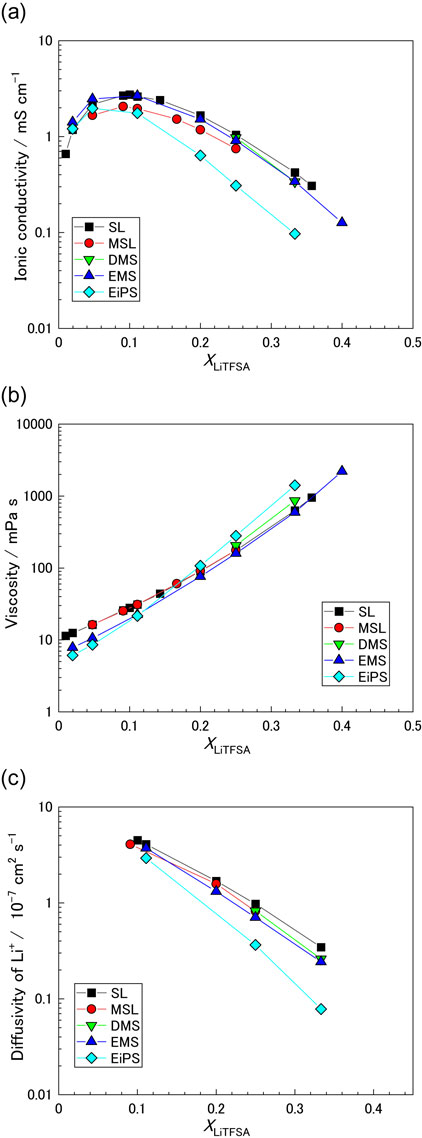
At high-salt concentration (XLiTFSA ≥ 0.2 and [LiTFSA]/[sulfone] ≥ 1/4), the viscosity of the EiPS-based electrolyte was slightly higher than that of other sulfone-based electrolytes, despite the low viscosity of bulky EiPS in low concentration region. This significant increase in viscosity is possibly because the large molecular size of EiPS providing larger solvate ion ([Li(EiPS)n]+) compared with other sulfone solvents. The ionic conductivities of the electrolytes with different sulfones were in the following order: SL > DMS ∼ EMS > MSL > EiPS. This trend is consistent with that observed in the LiBF4/sulfone systems in a previous study.20 The low conductivity of the EiPS-based electrolyte could have been caused by its high viscosity. The SL-based electrolyte exhibited a slightly higher viscosity than the EMS-based electrolyte at the same mole fraction XLiTFSA (Fig. 4b and Tables S2–S5). However, the SL-based electrolyte exhibited a higher ionic conductivity than the EMS-based electrolyte (Fig. 4a). This can be attributed to the higher diffusivity of Li+ in the SL electrolyte (Fig. 4c).
To further investigate the ion transport mechanism, the diffusivities of sulfone (Dsol) and the TFSA anions (DTFSA) were also measured (Tables S2–S5). Figure 5 shows the diffusivity ratios, DLi/Dsol and DLi/DTFSA, in sulfone-based electrolytes as a function of XLiTFSA. The Li+ cation is solvated by sulfone molecules ([Li(sulfone)n]+). In electrolytes with mole fractions of XLiTFSA < 0.2 ([LiTFSA]/[sulfone] < 1/4), coordinated and uncoordinated sulfone molecules coexist. In fact, PFG-NMR cannot distinguish between the coordinated and uncoordinated (free) sulfone molecules because Li+ ions dynamically exchange the solvent molecules in their first solvation sheath. The residence time of a sulfone in the first solvation sheath of Li+ is shorter than the time scale of the NMR measurements.54 Consequently, the measured diffusivity of sulfone reflects the average value of the coordinated and free solvent molecules.55 The diffusivity of a species is inversely proportional to its hydrodynamic radius (Stokes radius) in a continuous medium; therefore, the large solvated Li+ ion ([Li(solvent)n]+) exhibits a lower diffusivity than the free solvent.40,42,55,56 This results in a smaller DLi compared to Dsol in the solvent-excess electrolytes of XLiTFSA < 0.2 ([LiTFSA]/[sulfone] < 1/4), as shown in Fig. 5a. DLi/Dsol increased with increasing salt concentration in all solutions because the number of free sulfone molecules decreased. In the case of SL-based electrolytes, DLi/Dsol exceeded unity in the high salt concentration range of XLiTFSA ≥ 0.25 ([LiTFSA]/[sulfone] ≥ 1/3), suggesting that Li+ ions diffuse faster than SL. Figure 5b shows the DLi/DTFSA values in sulfone-based electrolytes. In the solvent-excess electrolytes of XLiTFSA ∼ 0.1, the majority of TFSA anions were free (Fig. 3). The hydrodynamic radius of the free TFSA anion is smaller than that of the solvated Li+, and TFSA diffuses faster than Li+. Upon increasing the LiTFSA concentration, the population of CIPs and AGGs increased, and the fraction of TFSA anions involved in Li+ coordination increased. PFG-NMR cannot distinguish between free and coordinated TFSA anions because the Li+ ions dynamically exchange them, i.e., the measured diffusivity of TFSA is the average value of the free and coordinated TFSA anions. DLi/DTFSA also increases with increasing salt concentration because of the increase in the population of CIPs and AGGs in the electrolytes. Except for the EiPS-based electrolyte, the DLi/DTFSA values of all the high-salt-concentration electrolytes exceeded unity at XLiTFSA = 0.33 ([LiTFSA]/[sulfone] = 1/2), indicating that Li+ ions diffuse faster than anions. It is apparent that the Li+ ion transport mechanism in highly concentrated electrolytes is different from that in low-salt concentration electrolytes.

In highly concentrated sulfone-based electrolytes, a single sulfone molecule is coordinated to different Li+ ions,18 and a sulfone-bridged structure, Li+–sulfone–Li+, is formed. Furthermore, in AGGs formed at high salt concentrations, a single TFSA anion is coordinated to multiple Li+ ions to produce an anion-bridged Li+–TFSA–Li+ structure. As previously mentioned, Li+ ions dynamically exchange ligands (sulfone molecules and TFSA anions) in their first solvation sheath. In high-concentration electrolytes, different solvated Li+ ions and AGGs are adjacent to each other. In certain highly concentrated electrolytes, Li+ ions move faster than the ligands by exchanging ligands, i.e., Li+ hopping conduction occurs, as previously reported.18–20,25 The structure of the sulfone solvent apparently affects the Li+ ion conduction process. For the [LiTFSA]/[sulfone] = 1/2 electrolyte, both DLi/Dsol and DLi/DTFSA exhibited the following order: SL > DMS > EMS > EiPS (Fig. 5). Except for the SL-based system, both DLi/Dsol and DLi/DTFSA increased with decreasing the molecular size of sulfone solvent, suggesting that smaller sulfone is favorable for enhancing the Li+ ion transport. The volumetric LiTFSA concentrations in EiPS, which has a large molecular size, were relatively lower than those in other sulfone-based electrolytes at similar LiTFSA mole fractions (Fig. S2); therefore, the average distance between the Li+ ions is longer in EiPS-based electrolytes than in other sulfone-based electrolytes. Furthermore, the ethyl and isopropyl groups of EiPS may sterically hinder the ligand exchange of the Li+ ions bound to the SO2 groups of EiPS.20 SL-based electrolytes exhibited higher DLi/Dsol and DLi/DTFSA values than other sulfone-based electrolytes. SL is a five-membered cyclic compound, and the energy barriers for the conformational change (pseudorotation) of the five membered-ring is low as well as the case of cyclopentane.57,58 The facile conformational change of SL might enhance the ligand-exchange of Li+. This hypothesis is further supported by relatively high DLi/Dsol and DLi/DTFSA in MSL-based electrolytes (Fig. 5) despite the relatively low volumetric LiTFSA concentration (Fig. S2). Overall, sulfone solvents with compact molecular sizes and low energy barriers of conformational change are favorable for enhancing Li+ hopping conduction in highly concentrated electrolytes. Molecular dynamics simulations for the [LiTFSA]/[sulfone] = 1/2 electrolytes are underway in our group to further understand the Li+ ion transport mechanism and will be reported in the forthcoming paper.
Finally, the effects of the Li+ hopping conduction on the Li+ transference number (tLi+abc) were examined under anion-blocking conditions. The value of tLi+abc was evaluated using a symmetric Li/Li cell (Fig. S3 and Table S6). The tLi+abc values of the [LiTFSA]/[sulfone] = 1/2 electrolytes are listed in Table 1. The tLi+abc values for SL- and DMS-based electrolytes were 0.70 and 0.69, respectively, while those for EMS- and EiPS-based electrolytes were 0.53 and 0.37, respectively. For comparison, the Li+ transference numbers calculated from the diffusivities of Li+ and TFSA− (tLi+NMR = DLi/(DLi + DTFSA)) are shown in Table 1. The tLi+abc value of each electrolyte was different from its tLi+NMR value. This is because tLi+NMR was calculated without considering the dynamic correlative motion of the ions in the electrolyte. In an ideal dilute solution (where the Li salt is completely dissociated), tLi+NMR and tLi+abc should exhibit identical values. However, in concentrated electrolytes, correlative and anti-correlative motions of ions occur, which significantly affect the ion transport process.59–62 The tLi+abc value is the current fraction carried by Li+ to the ohmic current calculated from the potential difference across the bulk electrolyte and ionic conductivity; this value includes the effects of the dynamic correlative motions of ions. Therefore, the product of tLi+abc and ionic conductivity (σ), tLi+abc × σ, is a useful measure for evaluating the Li+ ion transport ability of an electrolyte under anion-blocking conditions, such as in Li-ion and Li–S batteries. As shown in Table 1, the [LiTFSA]/[SL] = 1/2 electrolyte exhibited the highest ionic conductivity and the highest tLi+abc, suggesting that SL is superior to other sulfone solvents in producing a highly concentrated electrolyte with high Li+ transport capability.
Table 1. LiTFSA concentrations (
cLi), viscosities (η), ionic conductivities (σ), diffusivity of Li
+ (
DLi), Li
+ transference numbers (
tLi+NMR and
tLi+abc), and Li
+ ion conductivities (
tLi+abc × σ) of the [LiTFSA]/[sulfone] = 1/2 electrolytes at 30 °C. Except for
tLi+abc, the data for the [LiTFSA]/[SL] = 1/2 solution were obtained from a previous report.
19 Notably, the [LiTFSA]/[MSL] = 1/2 mixture was solid at room temperature.
| sulfone |
cLi
(mol dm−3) |
η
(mPa s) |
σ
(mS cm−1) |
DLi
(10−7 cm2 s−1) |
tLi+NMR |
tLi+abc |
tLi+abc × σ
(mS cm−1) |
| SL |
2.97 |
627 |
0.42 |
0.35 |
0.61 |
0.70 |
0.30 |
| DMS |
3.36 |
861 |
0.34 |
0.26 |
0.58 |
0.69 |
0.23 |
| EMS |
3.01 |
592 |
0.34 |
0.24 |
0.53 |
0.53 |
0.18 |
| EiPS |
2.52 |
1409 |
0.01 |
0.08 |
0.45 |
0.37 |
0.04 |
4. Conclusions
The phase behavior and transport properties of binary mixtures containing LiTFSA and various sulfone solvents were analyzed in this study. Mixtures of LiTFSA and the sulfone solvents had low liquidus line temperatures and maintained a liquid state at room temperature over a wide composition range; however, certain sulfones formed stoichiometric crystalline solvates of LiTFSA: [LiTFSA]/[SL] = 1/1, [LiTFSA]/[MSL] = 1/1, and [LiTFSA]/[EiPS] = 1/1. The viscosity and ionic conductivity of the liquid-state mixtures increased and decreased, respectively, with increasing LiTFSA mole fraction. In highly concentrated liquid electrolytes, solvent-bridged (Li+–sulfone–Li+) and anion-bridged (Li+–TFSA–Li+) network structures were formed. We also observed that Li+ ions diffuse by exchanging ligands (sulfone and TFSA). The ratios of diffusivities DLi/Dsol and DLi/DTFSA increased with increasing mole fractions of LiTFSA in the electrolytes. Except for the EiPS-based electrolyte, Li+ ions diffuse faster than the TFSA anions in all electrolytes with a composition of [LiTFSA]/[sulfone] = 1/2, resulting in high Li+ transference numbers. The DMS- and SL-based electrolytes exhibited higher DLi/Dsol and DLi/DTFSA values than other sulfone-based electrolytes. Consequently, sulfone solvents with compact molecular sizes and low energy barriers of conformational change are favorable for enhancing the ligand exchange of Li+ ions in highly concentrated electrolytes. SL-based electrolytes show higher ionic conductivity and higher tLi+abc × σ values than other sulfone-based electrolytes, suggesting that SL is superior to other sulfone solvents in producing a highly concentrated electrolyte having a high Li+ transport capability.
Data Availability Statement
- 1)
Highly concentrated Li salt/aprotic solvent solutions are promising electrolytes for next-generation batteries. Understanding the Li+ ion transport process is crucial for designing novel battery electrolytes. In this study, we systematically investigated the phase behavior, solvate structures, and Li+ transport properties of binary mixtures comprising lithium bis(trifluoromethanesulfonyl)amide (LiTFSA) and various sulfones, such as sulfolane (SL), 3-methyl sulfolane (MSL), dimethyl sulfone (DMS), ethyl methyl sulfone (EMS), and ethyl isopropyl sulfone (EiPS). Except for the MSL system, the [LiTFSA]/[sulfone] = 1/2 mixtures remained in a liquid state at room temperature, thus enabling a systematic comparison of the Li+ transport properties in the highly concentrated electrolytes. In highly concentrated liquid electrolytes, Li+ ions diffuse by exchanging ligands (sulfone and TFSA). Li+ ions diffuse faster than TFSA in all electrolytes except the EiPS-based electrolyte at a composition of [LiTFSA]/[sulfone] = 1/2, resulting in high Li+ transference numbers. SL-based electrolytes show higher ionic conductivity and Li+ transference numbers than other sulfone-based electrolytes. Consequently, sulfone solvents with compact molecular sizes and low energy barriers of conformational change are favorable for enhancing the Li+ ion transport in the electrolytes.
CRediT Authorship Contribution Statement
Ryoichi Tatara: Investigation (Lead), Validation (Lead), Writing – original draft (Lead)
Yosuke Ugata: Investigation (Supporting), Validation (Supporting), Writing – original draft (Supporting)
Shuhei Miyazaki: Investigation (Supporting)
Natsuki Kishida: Investigation (Supporting)
Shohei Sasagawa: Investigation (Supporting)
Kazuhide Ueno: Validation (Supporting)
Seiji Tsuzuki: Validation (Supporting)
Masayoshi Watanabe: Funding acquisition (Lead), Validation (Supporting)
Kaoru Dokko: Funding acquisition (Lead), Supervision (Lead), Writing – review & editing (Lead)
Conflict of Interest
The authors declare no conflicts of interest.
Funding
Japan Society for the Promotion of Science: JP19H05813
Japan Society for the Promotion of Science: JP21H04697
Japan Society for the Promotion of Science: JP22H00340
Advanced Low Carbon Technology Research and Development Program: JPMJAL1301
Footnotes
R. Tatara: Present address: Department of Applied Chemistry, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku, Tokyo 162-8601, Japan
R. Tatara, Y. Ugata, K. Ueno, and K. Dokko: ECSJ Active Members
S. Miyazaki: ECSJ Student Member
M. Watanabe: ECSJ Fellow
References
- 1) K. Xu, Chem. Rev., 104, 4303 (2004).
- 2) K. Xu, Chem. Rev., 114, 11503 (2014).
- 3) M. Gauthier, T. J. Carney, A. Grimaud, L. Giordano, N. Pour, H. H. Chang, D. P. Fenning, S. F. Lux, O. Paschos, C. Bauer, F. Maglia, S. Lupart, P. Lamp, and Y. Shao-Horn, J. Phys. Chem. Lett., 6, 4653 (2015).
- 4) K. Izutsu, Electrochemistry in Nonaqueous Solutions (2nd ed.), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim (2009).
- 5) M. S. Ding, K. Xu, S. S. Zhang, K. Amine, G. L. Henriksen, and T. R. Jow, J. Electrochem. Soc., 148, A1196 (2001).
- 6) K. Dokko, Electrochemistry, 90, 101003 (2022).
- 7) Y. Ugata, K. Shigenobu, R. Tatara, K. Ueno, M. Watanabe, and K. Dokko, Phys. Chem. Chem. Phys., 23, 21419 (2021).
- 8) Y. Yamada, J. Wang, S. Ko, E. Watanabe, and A. Yamada, Nat. Energy, 4, 269 (2019).
- 9) S. Ko, T. Obukata, T. Shimada, N. Takenaka, M. Nakayama, A. Yamada, and Y. Yamada, Nat. Energy, 7, 1217 (2022).
- 10) T. Tonoya, H. Yamamoto, Y. Matsui, H. Hinago, and M. Ishikawa, Electrochemistry, 90, 107003 (2022).
- 11) H. Miyauchi, K. Inaba, K. Takahashi, N. Arai, Y. Umebayashi, and S. Seki, Sustain. Energy Fuels, 6, 4218 (2022).
- 12) N. Arai, H. Watanabe, E. Nozaki, S. Seki, S. Tsuzuki, K. Ueno, K. Dokko, M. Watanabe, Y. Kameda, and Y. Umebayashi, J. Phys. Chem. Lett., 11, 4517 (2020).
- 13) G. Kamesui, K. Nishikawa, M. Ueda, and H. Matsushima, ACS Energy Lett., 7, 4089 (2022).
- 14) T. Doi, R. Masuhara, M. Hashinokuchi, Y. Shimizu, and M. Inaba, Electrochim. Acta, 209, 219 (2016).
- 15) K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko, and M. Watanabe, J. Am. Chem. Soc., 133, 13121 (2011).
- 16) Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama, and A. Yamada, J. Am. Chem. Soc., 136, 5039 (2014).
- 17) K. Takada, Y. Yamada, E. Watanabe, J. Wang, K. Sodeyama, Y. Tateyama, K. Hirata, T. Kawase, and A. Yamada, ACS Appl. Mater. Interfaces, 9, 33802 (2017).
- 18) K. Dokko, D. Watanabe, Y. Ugata, M. L. Thomas, S. Tsuzuki, W. Shinoda, K. Hashimoto, K. Ueno, Y. Umebayashi, and M. Watanabe, J. Phys. Chem. B, 122, 10736 (2018).
- 19) A. Nakanishi, K. Ueno, D. Watanabe, Y. Ugata, Y. Matsumae, J. Liu, M. L. Thomas, K. Dokko, and M. Watanabe, J. Phys. Chem. C, 123, 14229 (2019).
- 20) Y. Ugata, S. Sasagawa, R. Tatara, K. Ueno, M. Watanabe, and K. Dokko, J. Phys. Chem. B, 125, 6600 (2021).
- 21) Y. Ugata, Y. Chen, S. Sasagawa, K. Ueno, M. Watanabe, H. Mita, J. Shimura, M. Nagamine, and K. Dokko, J. Phys. Chem. C, 126, 10024 (2022).
- 22) Y. Okamoto, S. Tsuzuki, R. Tatara, K. Ueno, K. Dokko, and M. Watanabe, J. Phys. Chem. C, 124, 4459 (2020).
- 23) R. Tatara, Y. Okamoto, Y. Ugata, K. Ueno, M. Watanabe, and K. Dokko, Electrochemistry, 89, 590 (2021).
- 24) Y. Ugata, R. Tatara, K. Ueno, K. Dokko, and M. Watanabe, J. Chem. Phys., 152, 104502 (2020).
- 25) Y. Ugata, M. L. Thomas, T. Mandai, K. Ueno, K. Dokko, and M. Watanabe, Phys. Chem. Chem. Phys., 21, 9759 (2019).
- 26) S. Kondou, M. L. Thomas, T. Mandai, K. Ueno, K. Dokko, and M. Watanabe, Phys. Chem. Chem. Phys., 21, 5097 (2019).
- 27) S. Kondou, K. Dokko, M. Watanabe, and K. Ueno, Electrochemistry, 89, 389 (2021).
- 28) R. Tatara, S. Nishimura, Y. Okamoto, K. Ueno, M. Watanabe, and K. Dokko, J. Phys. Chem. C, 124, 15800 (2020).
- 29) K. M. Diederichsen, E. J. McShane, and B. D. McCloskey, ACS Energy Lett., 2, 2563 (2017).
- 30) M. D. Galluzzo, J. A. Maslyn, D. B. Shah, and N. P. Balsara, J. Chem. Phys., 151, 020901 (2019).
- 31) N. Tachikawa, K. Yamauchi, E. Takashima, J. W. Park, K. Dokko, and M. Watanabe, Chem. Commun., 47, 8157 (2011).
- 32) K. Dokko, N. Tachikawa, K. Yamauchi, M. Tsuchiya, A. Yamazaki, E. Takashima, J. W. Park, K. Ueno, S. Seki, N. Serizawa, and M. Watanabe, J. Electrochem. Soc., 160, A1304 (2013).
- 33) S. S. Zhang, J. Power Sources, 231, 153 (2013).
- 34) F. Y. Fan, W. C. Carter, and Y. M. Chiang, Adv. Mater., 27, 5203 (2015).
- 35) C. Zhang, A. Yamazaki, J. Murai, J.-W. Park, T. Mandai, K. Ueno, K. Dokko, and M. Watanabe, J. Phys. Chem. C, 118, 17362 (2014).
- 36) E. V. Karaseva, L. A. Khramtsova, A. N. Lobov, E. V. Kuzmina, D. Eroglu, and V. S. Kolosnitsyn, J. Power Sources, 548, 231980 (2022).
- 37) G. G. Eshetu, X. Judez, C. Li, M. Martinez-Ibanez, I. Gracia, O. Bondarchuk, J. Carrasco, L. M. Rodriguez-Martinez, H. Zhang, and M. Armand, J. Am. Chem. Soc., 140, 9921 (2018).
- 38) G. M. Sheldrick, Acta Crystallogr. A Found. Adv., A71, 3 (2015).
- 39) G. M. Sheldrick, Acta Crystallogr. C Struct. Chem., C71, 3 (2015).
- 40) K. Ueno, R. Tatara, S. Tsuzuki, S. Saito, H. Doi, K. Yoshida, T. Mandai, M. Matsugami, Y. Umebayashi, K. Dokko, and M. Watanabe, Phys. Chem. Chem. Phys., 17, 8248 (2015).
- 41) H. Tokuda, K. Hayamizu, K. Ishii, M. A. B. H. Susan, and M. Watanabe, J. Phys. Chem. B, 108, 16593 (2004).
- 42) C. Zhang, K. Ueno, A. Yamazaki, K. Yoshida, H. Moon, T. Mandai, Y. Umebayashi, K. Dokko, and M. Watanabe, J. Phys. Chem. B, 118, 5144 (2014).
- 43) M. M. Gafurov, K. S. Rabadanov, V. D. Prisyazhnyi, D. O. Tret’Yakov, M. I. Gorobets, S. A. Kirillov, M. B. Ataev, and M. M. Kakagasanov, Russ. J. Phys. Chem. A, 85, 1499 (2011).
- 44) R. Tatara, D. G. Kwabi, T. P. Batcho, M. Tulodziecki, K. Watanabe, H. M. Kwon, M. L. Thomas, K. Ueno, C. V. Thompson, K. Dokko, Y. Shao-Horn, and M. Watanabe, J. Phys. Chem. C, 121, 9162 (2017).
- 45) R. Tatara, G. M. Leverick, S. Feng, S. Wan, S. Terada, K. Dokko, M. Watanabe, and Y. Shao-Horn, J. Phys. Chem. C, 122, 18316 (2018).
- 46) K. Fujii, H. Hamano, H. Doi, X. Song, S. Tsuzuki, K. Hayamizu, S. Seki, Y. Kameda, K. Dokko, M. Watanabe, and Y. Umebayashi, J. Phys. Chem. C, 117, 19314 (2013).
- 47) Y. Umebayashi, T. Mitsugi, K. Fujii, S. Seki, K. Chiba, H. Yamamoto, J. N. Canongia Lopes, A. A. Padua, M. Takeuchi, R. Kanzaki, and S. Ishiguro, J. Phys. Chem. B, 113, 4338 (2009).
- 48) A. K. Chan, R. Tatara, S. Feng, P. Karayaylali, J. Lopez, I. E. L. Stephens, and Y. Shao-Horn, J. Electrochem. Soc., 166, A1867 (2019).
- 49) W. A. Henderson, M. L. Helm, D. M. Seo, P. C. Trulove, H. C. De Long, and O. Borodin, J. Electrochem. Soc., 169, 060515 (2022).
- 50) Y. Yamada, Y. Takazawa, K. Miyazaki, and T. Abe, J. Phys. Chem. C, 114, 11680 (2010).
- 51) J. M. Alía and H. G. M. Edwards, Vib. Spectrosc., 24, 185 (2000).
- 52) M. Yamagata, N. Tachikawa, Y. Katayama, and T. Miura, Electrochim. Acta, 52, 3317 (2007).
- 53) R. Tatara, K. Ueno, K. Dokko, and M. Watanabe, ChemElectroChem, 6, 4444 (2019).
- 54) C. Zhang, K. Ueno, A. Yamazaki, K. Yoshida, H. Moon, T. Mandai, Y. Umebayashi, K. Dokko, and M. Watanabe, J. Phys. Chem. B, 118, 5144 (2014).
- 55) K. Hayamizu, Y. Aihara, S. Arai, and C. G. Martinez, J. Phys. Chem. B, 103, 519 (1999).
- 56) K. Ueno, K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko, and M. Watanabe, J. Phys. Chem. B, 116, 11323 (2012).
- 57) H. M. Badawi, W. Forner, B. El Ali, and A. R. Al-Durais, Spectrochim. Acta, Part A, 70, 983 (2008).
- 58) E. J. Ocola, L. E. Bauman, and J. Laane, J. Phys. Chem. A, 115, 6531 (2011).
- 59) N. M. Vargas-Barbosa and B. Roling, ChemElectroChem, 7, 367 (2020).
- 60) D. Dong, F. Salzer, B. Roling, and D. Bedrov, Phys. Chem. Chem. Phys., 20, 29174 (2018).
- 61) K. Shigenobu, K. Dokko, M. Watanabe, and K. Ueno, Phys. Chem. Chem. Phys., 22, 15214 (2020).
- 62) K. Shigenobu, M. Shibata, K. Dokko, M. Watanabe, K. Fujii, and K. Ueno, Phys. Chem. Chem. Phys., 23, 2622 (2021).

