Review
MECHANISTIC INSIGHTS OF CORONARY VASOSPASM AND NEW THERAPEUTIC APPROACHES
2015 Volume 61 Issue 1 Pages 1-12

Details
2015 Volume 61 Issue 1 Pages 1-12
Coronary vasospasm is a major health problem in Japan because of its high incidence and severity. Great advances have been made in clinical research and therapy focusing on coronary vasospastic angina. In particular, the Ca antagonist has contributed to improvements in the quality of life of patients with coronary vasospastic angina. Moreover, several animal models of coronary vasospasm have provided insight into the pathophysiology of this disease. However, the mechanisms of coronary vasospasm and microvascular spasm remain elusive. We have studied the mechanisms of coronary vasospasm, including microvascular spasm, in an animal model. The present study was designed to develop an animal model of coronary vasospasm and to elucidate the mechanisms and pathophysiology of coronary vasospasm and new therapeutic approaches. The topics discussed in this article include: (1) the role of endothelial and smooth muscle function on the genesis of coronary vasospasm, (2) the participation of reactive oxygen species in coronary vasospasm, and (3) aging and spasm. We hope that the contents of this volume will be useful to “students of coronary vasospasm” at many levels, including the clinician involved in cardiovascular studies and scientists in various disciplines who are interested in a comprehensive, in-depth review of coronary vasospasm.
Coronary vasospasm can be defined as a reversible, focal, and intense coronary vasoconstriction that typically occurs in an epicardial conduit artery segment. The definitive diagnosis of a coronary artery spasm is determined by the direct visualization of such a reversible focal subtotal coronary occlusion at a site distal to the catheter tip. This is generally associated with regional ST segment elevation and angina. The first description of coronary artery spasm was likely made by Heberden in 1802 when he described angina as spasmodic, occurring at rest and related to emotional factors.1) For over 100 years, this presumed vasoconstrictor phenomenon was implicated in myocardial infarction and angina pectoris. It was not until the mid-twentieth century that the correlation between atherosclerotic plaques and angina pectoris was realized.2) This observation lessened the interest in vasospasms as an etiology of angina, particularly because later investigations failed to observe spasms very often in patients with angina pectoris.3) In the latter third of the twentieth century, the demonstration of coronary vasospasm during angina (Prinzmetal’s angina) and as a precursor to myocardial infarction4) resurrected interest in the vasomotor etiology of angina pectoris.5) In recent years, our understanding of the endothelial modulation of coronary tone has evolved, and substantial insight has been provided from a number of studies over the past three decades. However, the mechanism of coronary vasospasm remains elusive.
Some investigations have reported that endothelial dysfunction is present at the site of a coronary vasospasm.6,7) However, several lines of evidence indicate that coronary vasospasm is caused primarily by smooth muscle hypercontraction whereas the contribution of endothelial dysfunction may be minimal.8) Thus, the role of endothelial dysfunction in the mechanism of coronary vasospasm remains controversial. Recently, several animal models of coronary vasospasm have provided some insight into the pathophysiology of this disease.9) The method of inducing coronary spasm in animals reflects our understanding of the underlying pathophysiology. Shimokawa et al. have developed a porcine model of coronary vasospasm with atherosclerosis through a combination of endothelium removal and high-cholesterol feeding.10) One month following endothelial removal, histamine evokes a potent coronary vasospasm. This site of endothelial removal and spasm shows focal intimal thickening, similar to what has been observed using intravascular ultrasound in humans.11,12) Takeshita and Shimokawa et al. have developed a porcine model of focal coronary vasospasm in which the coronary adventitia is chronically exposed to interleukin-1β.13,14) The subsequent application of serotonin produces focal vasospasm. However, dilations to A23187, substance P, and bradykinin were normal, suggesting that endothelial function was normal. Thus, endothelial dysfunction is not required in this model of coronary vasospasm. This model takes advantage of the correlation between inflammation and coronary vasospasm and suggests a possible mechanistic link. Hermsmeyer et al. developed a primate model of coronary spasm that was dependent upon sex hormone levels and did not require endothelial denudation or atherosclerosis.15,16) Studies have also been performed on ovariectomized rhesus monkeys. It was found that the combination of a pathophysiological concentration of serotonin and the thromboxane A2 mimetic U46619 injected through an intracoronary catheter synergistically caused coronary vasospasm. These drug-induced spasms were similar to vasospasms induced by mechanical vascular injury followed by serotonin and to those stimulated in human intracoronary diagnostic tests. The induced spasms were prevented by treatment with progesterone, indicating that natural ovarian steroids reduce hyperreactivity to serotonin. A reduction in intracellular calcium and protein kinase C levels was also reported in these rhesus monkeys’ spasm.17) The specific type of hormone replacement was demonstrated to be important. Treatment with estrogen or progesterone effectively prevented the induction of spasm. These studies indicate that coronary vasospasm can be stimulated without preexisting vascular pathology, endothelial denudation, or injury, and have implications for hormone supplementation in postmenopausal women. Other animal models have been used as well. Vasopressin and endothelin induce myocardial ischemia in conscious rabbits and in rats, presumably via coronary spasm.18,19) Intracoronary methacholine also precipitates spasms in rats.20) Cardiomyopathic hamsters experiencing chronic stress may also demonstrate coronary spasms in response to vasopressin.21) X-irradiation can induce coronary spasms in porcine coronary segments following denudation.22) However, these coronary vasospasms mentioned above are not spontaneous vasospasms but drug-induced ones. In addition, coronary vasospasm is likely to be the result of a complex constellation of abnormalities that are difficult to mimic in animal models; there may be species variability as well. Thus, a novel model of spontaneous coronary vasospasm may help explain the etiology of this disease. We have developed such a model in the conscious and unrestrained state using repeated coronary artery endothelial injury,23,24) which may be linked to vascular smooth muscle hypercontraction; this type of injury is likely to occur repeatedly over the course of a lifetime as a result of local shear forces and various risk factors (such as hypercholesterolemia, diabetes mellitus, and hypertension).9) Using this original spasm model, we investigated the mechanism of coronary vasospasm in which impaired endothelial functioning leads to heightened vascular smooth muscle reactivity and provokes intense conduit coronary artery constriction; we also examined the possibility of therapeutic agents such as antioxidants, β1-blockers, and endothelin type-A antagonist25-28) as therapeutic approaches.
From recent studies, in some patients without epicardial coronary obstruction, the pharmacological provocation of coronary spasm produces a reduction in coronary flow without conduit artery constriction.29-31) The reduction in flow is promptly restored by vasodilator therapy, indicating its reversibility. This phenomenon has been termed microvascular spasm. Calcium channel blockers are less effective in alleviating symptoms of microvascular spasm,32,33) and the pathological processes involved in microvascular spasm are unclear; whether this phenomenon represents a distinct clinical entity is also unknown. One reason for the lack of progress is that a microvascular spasm model is not available. We hypothesized that microvascular spasm is caused by a diffuse increase in arteriolar sensitivity to vasoconstrictor stimuli because of upstream epicardial coronary artery endothelial dysfunction, and we confirmed that the repeated endothelial injuries of the epicardial coronary artery induced not only spontaneous conduit coronary artery spasm but also downstream microvascular spontaneous spasm.23)
We hypothesized that coronary artery endothelial injury and regeneration is likely to occur repeatedly over the course of a lifetime as a result of local shear forces and various risk factors such as atherosclerosis, hypertension, hyperlipidemia, and tobacco smoke, or as a result of percutaneous transmural coronary angioplasty; therefore, we constructed a balloon endothelial injury pig model in the epicardial left anterior descending coronary artery every two weeks24) (Fig. 1). In this model, coronary artery endothelium was regenerated after two weeks of balloon injury with intimal hyperplasia. In the regenerated endothelium site, serotonin induced coronary artery spasm, and we observed a reduction in eNOS protein levels (Figs. 2 and 3). In the genesis of coronary vasospasm in this site, the decrease of endothelium-dependent vasodilation was primarily evaluated compared to the hypercontractility of the vascular smooth muscle. We have reported that hypercholesterolemia contributes to coronary vasospasm in this model; however, this risk factor appears to have been rarely reported in cases of Japanese human coronary vasospasm.34) Next, we examined whether coronary microvascular spasm occurs in this model and the role of platelets in microvascular spasm by measuring coronary blood flow and platelet function.35) From this study, epicardial repeated endothelial injuries were shown to induce not only vasospasm at the injured site but also downstream microvascular spasm with vessel remodeling. Additionally, thromboxane A2 as the product of platelet activation plays an important role in the etiology of microvascular spasm. The chronic suppression of platelet activation by the administration of aspirin with the thromboxane A2 synthase inhibitor appears to be effective in preventing microvascular spasm. However, this spasm that occurred in either the microvessel or the epicardial coronary artery was produced using vasoactive agents such as acetylcholine or serotonin, but it was not produced spontaneously. Thus, we hypothesized that repeated endothelial injuries in the large coronary artery cause spontaneous spasm in both the injured large coronary artery and its downstream microvessels. To verify this hypothesis, we observed coronary hemodynamics and cardiac surface electrocardiograms continuously using the telemetry system in this model (Fig. 1).23) Through this examination, we found that this model is suitable as a global spontaneous coronary artery spasm model. This model induces coronary vasospasm in the conscious state without any provocations, including drugs (Fig. 4). In this model, eNOS levels were globally reduced not only at the epicardial denuded site but also at the downstream peripheral site; additionally, the chronic administration of an antioxidant such as vitamin C or reduced glutathione is effective in preserving eNOS levels (Fig. 5). Moreover, the generation of superoxide and endothelin-1 increased in the epicardial spasm site (Fig. 6). In the spasm site of the epicardial coronary artery, myosin light-chain phosphorylation was induced by treatment with serotonin, which was inhibited by W-7, a calmodulin inhibitor (Fig. 7).

We performed coronary angiography with or without the intracoronary administration of serotonin (Se) or acetylcholine (ACh) using a 6 F. handmade Judkins type catheter for measuring the left anterior descending coronary artery (LAD) diameter with simultaneously recordings of electrocardiograms (ECG), aortic (Ao) blood pressure, and the change of LAD blood flow in anesthetized pigs. Next, balloon denudation was performed in the LAD just distal to the bifurcation of the LAD and the left circumflex coronary artery using a 2F Fogarty catheter. These procedures were performed every 2 weeks (W). We also observed hemodynamics and cardiac ischemic events under the conscious state with a telemetry system in the pigs.

Coronary spasm under in vivo conditions. Coronary angiography and electrocardiograms were obtained before and after the intracoronary administration of 10 µg/kg of serotonin at week 8 [2 weeks after 4 endothelial denudation (ED) procedures] in one example with a repeated ED in the left anterior descending (LAD) coronary artery group. LAD narrowing occurred at the ED site (arrow), corresponding with the ischemic ST-segment change in the electrocardiogram leads II and V5

Western blotting for endothelial nitric oxide synthase (eNOS) using the left anterior descending coronary artery (LAD) isolated samples at week 8 [2 weeks after 4 endothelial denudation (ED) procedures]. The results are shown as the means ± SEM, n=8 each, #; p<0.01 vs control group. The decrease of the eNOS protein levels was shown in the ED group (relative intensities: 0.25±0.05, p<0.01 vs control group) whereas it did not decrease in the coronary artery with chronic oral administration of the endothelin type-A receptor antagonist group (ED+ETA group, relative intensities: 0.76±0.08).

Representative tracing of the telemetry record in the pigs (A). Spontaneous coronary spasm with a decrease of coronary blood flow and ischemic ST-segment change occurred in the conscious pigs (B). ECG1, 2: electrocardiogram 1 (basal site of left ventricle), 2 (apex site of left ventricle) from cardiac surface leads. BP: blood pressure recorded from the catheter inserted in the intrathoratic artery, CBF: coronary blood flow in the left anterior descending coronary artery, which was recorded using a telemetric Doppler flow meter.

eNOS immunostaining at week 8 [2 weeks after 4 endothelial denudation (ED) procedures] in the repeated denuded portion and the downstream microvessel in ED, treatment with chronic oral administration of vitamin C or reduced glutathione, and in the corresponding portion of the control group. In the ED group, weak eNOS expression was observed in the endothelium of the denuded portion and the downstream microvessel. Antioxidants such as vitamin C or reduced glutathione have the preventive effect of eNOS down-regulation.

Expression of endothelin-1 (ET-1) and superoxide.
Representative images of ET-1 (a) and ECE (b) fluorescences in the isolated samples of the repeatedly denuded coronary artery sample (ED group) and the corresponding control artery sample at week 8 (2 weeks after 4 endothelial denudation procedures in the coronary artery). ET-1 fluorescence and protein levels of ET-1 determined using the ELISA method (c) increased in the ED group compared to those of the control group. ET-1 fluorescence augmented after the exposure of 1 µM serotonin in the ED group (a). ECE was slightly observed at week 8 in the control group whereas ECE increased at week 8 in the ED group (b). Moreover, dihydroethidium and P47phox fluorescence increased in the ED sample, which was augmented by the exposure of 1 µM serotonin in the ED group (d). The bar graphs are shown as the means ± SEM, n=8 each, #; p<0.01 vs control group in the same condition.

Myosin light chain phosphorylation.
Coronary vascular smooth muscle contraction originates from myosin light chain (MLC) phosphorylation in the media of the artery. We measured MLC phosphorylation using Western blotting after the exposure of 1 µM serotonin in the repeated endothelial denuded site (ED). W-7, CaM inhibitors and endothelin receptor-type A antagonists (ETA) inhibited the phosphorylation of MLC. However, hydroxyfasudil, Rho-kinase inhibitors partially changed the phosphorylation level of MLC. These data indicate that repeated endothelial denudation is the cause of serotonin-induced phosphorylation of the MLC via CaM in the coronary artery. The results are shown as the means ± SEM n=8 each, #; p<0.01 vs control group, *; p<0.01 vs ED group.
Aging associated with oxidant stress elevation is an important risk factor for the development of ischemic heart disease. In clinical settings, a shift in the redox equilibrium to a more oxidative state has been reported to occur in elderly patients with coronary artery spasm. SMP30 is a novel molecule whose expression decreases with age, and we have reported that SMP30 is closely related to the increase of oxidant stress that occurs with aging.36,37) In a previous SMP30 knockout mice study, we reported that chronic oxidant stress through NADPH oxidase activation with the SMP30 deficiency induced thiol oxidation in the coronary artery (Fig. 8) whereas attenuated bioactive nitric oxide generation resulted in coronary artery spasm in the heart (Figs. 9 and 10).38) Specifying the thiol redox state as a key regulator of eNOS activity provides a new platform for strategies to prevent coronary artery spasm and alleviate the underlying process of aging-induced coronary artery disease.

(a) The production of the superoxide anion radical (O2•−) and H2O2 reflect the staining with dihydroethidium (DHE) and dichlorodihydro-fluorescein fluorescence (DCF) in the vessel, respectively. We then examined reactive oxygen species (ROS) generation in the coronary arteries of the SMP30 KO mice based on the hypothesis that a SMP30 deficiency exacerbates ROS generation. The signals of the DHE and DCF staining were enhanced in the coronary arteries with a SMP30 deficiency. Apocynin, an NADPH oxidase inhibitor, decreased these signals in the coronary arteries of the SMP30 KO mice. These results suggest that superoxide generation in SMP30 deficiency depends on NADPH oxidase in the coronary arteries. (b) To assess thiol oxidation in the coronary arteries, the location of thiols was determined by the administration of fluorochromes monochlorobimane (MCB), which compounds covalently to react with reduced thiols. However, if the thiols are oxidized, the compounds do not bind. Acetylcholine (ACh) treatment decreased the fluorescence levels because of thiol oxidation in the coronary arteries of the wild type mice. The fluorescence levels because of MCB in the coronary arteries decreased in the SMP30 KO mice, and the level was restored with dithiothreitol (DTT), a reducing agent treatment. These results suggest that the total thiols were oxidized in the SMP30 KO mice. Elevation of the fluorescence of MCB by DTT indicates thiol reduction. ACh oxidized the intracellular thiols to a sufficient SMP30 level. A deficiency of SMP30 extinguishes additional thiol oxidation with ACh. Thus, reducing thiols might be essential for nitric oxide generation.
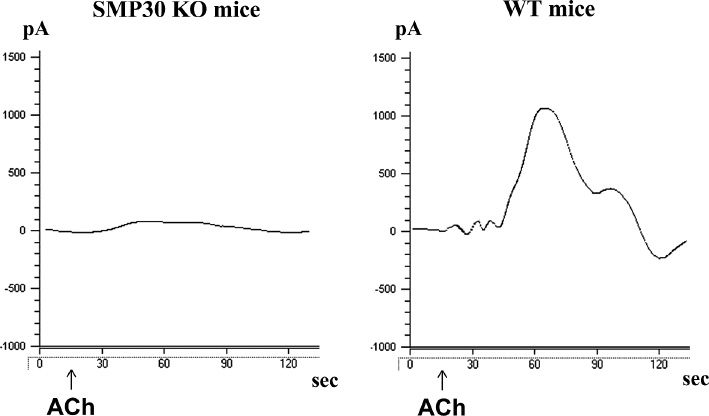
Nitric oxide (NO) generation in the aorta.
Representative electrochemical detection tracing NO concentrations in the isolated aorta from senescence marker protein-30 (SMP30) knockout mice and wild type mice in an organ chamber. The NO concentration is directly proportional to the current (pA), which was measured by a free radical analyzer (Apollo 4000). Acetylcholine-stimulated NO release was attenuated in SMP30 knockout mice vs. wild type mice (p<0.01). ACh indicates acetylcholine administration.

The intra-aortic sinus administration of acetylcholine (ACh, 2 µg) to SMP30 knockout mice induced transient ST-T segment elevation and reciprocal ST-T segment depression in the electrocardiograms. In the wild mice, the ACh-induced ST-T segment change did not appear. Before administration: before the administration of ACh, after 1, 2, and 5 min; 1, 2, and 5 min after the intra-aortic sinus administration of ACh.
The removal of precipitating factors is the first line of treatment for coronary spasm. Thus, all patients should be instructed to discontinue smoking, reduce alcohol intake, and eliminate any constrictor-provoking medications. The effectiveness of calcium channel blockers in alleviating the signs and symptoms of coronary spasm has resulted in this class of medications serving as the mainstay of therapy.39,40) In fact, a positive response of variant anginal symptoms to calcium blockers is considered both diagnostic and therapeutic. Both first- and second- generation agents in this class are effective. Nitrates are also effective for acute episodes, particularly in the catheterization laboratory, but are not as helpful for prophylaxis.41,42) The basis for the differential effectiveness of these 2 classes of drugs may be related to their effects on tonic and phasic coronary constriction. The periodic spontaneous constriction of the isolated cadaveric coronary artery occurs in patients in whom coronary disease is blocked by calcium channel blockers, but spontaneous tonic constriction is more sensitive to nitrates.43,44) The success with these 2 classes of agents either alone or in combination has minimized the necessity for additional therapeutic approaches. However, a recent report by Sueda et al.45) suggested that calcium channel blockers may not be as effective as previously determined because only 38% of patients on long-acting calcium channel blockers had complete resolution of symptoms. In addition to this therapeutic concern, a certain number of patients cannot tolerate these agents, and alternative approaches are required.
Nicorandil, which opens K-ATP channels and dilates the coronary artery, is often used in coronary vasospasm patients. It has been believed that the action of K-ATP channel opening drugs on the coronary artery is attributed mainly to the direct effect of these agents on the vascular K-ATP channel [called a nucleoside diphosphate-dependent K channel (K-NDP channel), exhibits properties significantly different from those of the cardiac K-ATP channel]. The cardiac K-ATP channel is the prototype of K-ATP channels possessing approximately 80 pS of single-channel conductance in the presence of approximately 150 mM extracellular K+ and opens spontaneously in the absence of ATPi. The K-NDP channel has the single-channel conductance of approximately 30-40 pS in the presence of approximately 150 mM extracellular K+, is closed in the absence of ATPi, and requires intracellular nucleoside di- or triphosphates, including ATPi to open. Nevertheless, K-ATP and K-NDP channels are both activated by K+ channel openers, including pinacidil and nicorandil, and inhibited by sulfonylurea derivatives such as glibenclamide. It recently was found that the cardiac K-ATP channel is composed of a sulfonylurea receptor (SUR) 2A and a two-transmembrane-type K+ channel subunit Kir6.2, while the vascular K-NDP channel may be the complex of SUR2B and Kir6.1. A number of K-ATP channel opening drugs have been developed in the last several decades that target ischemic heart disease and hypertension.46) Among these drugs, only nicorandil has been clinically proven to be effective in anti-anginal therapy, and many others cannot be adopted for clinical usage because of their significant side effects, including lower extremity edema.47) Kakkar et al. indicated the possibility that ex-smooth muscle mechanisms might be involved in the action of some K-ATP channel opening drugs, which might result in clinically different effectiveness.48) They tested the role of the K-NDP channel in spontaneous coronary vasospasm using the transgenic mice that harbored the K-NDP channel in only vascular smooth muscle cells but not any other cells. They observed not only protein expression but also the restoration of the functional K-NDP channel in the smooth muscle cells. These mice still exhibited vasospasm, abnormality in the ST-segment in an electrocardiogram, high coronary perfusion pressure, and sudden death. These results now clearly indicate the need for a reevaluation of the current consensus regarding the role of K-ATP channel opening drugs in coronary vasospasm.
Denopamine, a β1-selective agonist, has been demonstrated to markedly reduce the frequency and severity of attacks in a study of 10 patients with coronary vasospasm; complete elimination was achieved in 7 subjects.49) Further studies are required to confirm the utility of this treatment.
The antioxidant vitamin E improves endothelium-dependent vasodilation and reduces oxidative stress in subjects with coronary vasospasm; similarly, vitamin C displays these attributes, which was examined in our pig model.50) In subjects with coronary spasm, vitamin E levels are reduced.51) Further clinical studies are required to warrant the potential therapeutic benefits of antioxidants in patients with coronary vasospasm.
One of the exciting recent pharmacological approaches to coronary vasospasm is based on the mechanism of vascular smooth muscle hypercontractility. The increase in calcium sensitivity because of elevated levels of Rho-kinase is a feature specific for spastic segments of coronary arteries. The inhibition of Rho-kinase has been effective in reducing the ability to provoke spasm both in animal models and in humans.52,53) In a recent report, 20 patients with documented coronary artery spasm to intracoronary acetylcholine underwent a second challenge after receiving either intracoronary saline or a Rho-kinase inhibitor, fasudil, in a dose that had no effect on baseline hemodynamics.54) Fasudil reduced angiographic spasm and eliminated the associated chest pain and electrocardiographic changes. The results of this pilot study prompted an ongoing clinical trial of fasudil treatment of patients with coronary vasospasm.
Despite maximal medical therapy, a rare patient may have persistent vasospastic episodes. In these cases, proven therapeutic options do not exist. From our basic studies, we propose the possibility of the therapeutic effects of antioxidants and antiplatelets for coronary vasospasm. Whether these findings actually imply vascular protection and whether other potential protective factors against coronary events exist in patients with persistent vasospasm are challenging issues that should be addressed in future studies.
We thank Kiyoaki Katahira and my colleague for the animal care and experiments.