review
MEMBRANE TYPE 1-MATRIX METALLOPROTEINASE (MT1-MMP) IDENTIFIED AS A MULTIFUNCTIONAL REGULATOR OF VASCULAR RESPONSES
2015 年 61 巻 2 号 p. 91-100

詳細
2015 年 61 巻 2 号 p. 91-100
Membrane type 1-matrix metalloproteinase (MT1-MMP) functions as a signaling molecules in addition to a transmembrane metalloprotease, which degrades interstitial collagens and extracellular matrix components. This review focuses on the multifunctional roles of MT1-MMP as a signaling molecule in vascular responses to pro-atherosclerotic stimuli in the pathogenesis of cardiovascular diseases. First, the lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1)-MT1-MMP signaling axis contributes to endothelial dysfunction, which is mediated via small GTP-binding protein RhoA and Rac1 activation. Second, MT1-MMP plays a crucial role in reactive oxygen species (ROS) generation through the activation of receptor for advanced glycation end products (AGEs) in smooth muscle cells, indicating that MT1-MMP may be a therapeutic target for diabetic vascular complications. Third, MT1-MMP is involved in RhoA/Rac1 activation and Ca2+ signaling in the mechanism of thrombin-stimulated endothelial dysfunction and oxidant stress. Fourth, the inhibition of the MT1-MMP/Akt signaling pathway may be an attractive strategy for treating endothelial disordered hemostasis in the development of vascular diseases linked to TNF-α-induced inflammation. Fifth, MT1-MMP through RAGE induced RhoA/Rac1 activation and tissue factor protein upregulation through NF-κB phosphorylation in endothelial cells stimulated by high-mobility group box-1, which plays a key role in the systemic inflammation. These findings suggest that the MT1-MMP-mediated signaling axis may be a promising target for treating atherosclerosis and subsequent cardiovascular diseases.
Matrix metalloproteinase (MMP) and membrane-type MMPs (MT-MMPs), a large family of zinc-dependent endopeptidases, are the fibrinolysins responsible for degrading a variety of extracellular matrix (ECM) components1,2). MT-MMPs were identified as multifunctional enzymes for modulating the bioactivity of transmembrane receptors, which was anchored to the cell membrane instead of being soluble1-3). MT-MMPs have been well known to serve as important enzymes engaged by tumor cells in the mechanisms of metastasis4). All MT-MMPs act at the cell surface, and membrane type 1-MMP (MT1-MMP), an activator of pro-MMP-2, was localized predominantly in human invasive breast carcinomas and spontaneously metastasizing melanoma cell lines, indicating that MT1-MMP may play a key role in the promotion of tumor cell invasion, metastasis, and angiogenesis5,6). Degradation of the vascular ECM by MT-MMPs is critical for smooth muscle cell (SMC) migration, plaque instability, and consequent hypercoagulability in the pathogenesis of atherosclerosis and aortic aneurysms7,8). Recent studies of bone marrow transplantation in MT1-MMP2/2 mice have demonstrated that macrophage-derived MT1-MMP plays a role in the pathogenesis of plaque stability9,10). MT1-MMP may be a key MMP responsible for effecting postinfarction cardiac ECM remodeling and cardiac dysfunction11). MT-MMPs also play important roles for remodeling ECM during vascular injury, cell recruitment to the vessel wall, endothelial cell growth, and angiogenesis12,13). Proinflammatory molecules including tumor necrosis factor (TNF)-α and oxidized, low-density lipoprotein (ox-LDL) upregulate MT1-MMP expression in vascular SMCs and macrophages in human atherosclerotic plaques, and MT1-MMP activated by proinflammatory molecules affects ECM remodeling in the pathology of atherosclerosis14,15). MT1-MMP regulates MMP-2 expression and angiogenesis-related functions in human umbilical vein endothelial cells16). Rajavashisth et al. also reported that activation of endothelial cells by inflammatory cytokines and/or ox-LDL increases MT1-MMP expression17). Schneider et al. reported that MT1-MMP deficiency in bone marrow-derived cells promotes collagen accumulation in mouse atherosclerotic plaques18). To our knowledge, MT1-MMP affects various cellular functions not only as a pericellular protease but also as a signaling molecule in both physiological and pathological settings19-21).
Here, we focus our investigations on multifunctional roles of MT1-MMP as a signaling molecule in vascular responses to atherosclerotic stimuli and suggest that MT1-MMP-mediated signaling pathways investigated may be a therapeutic target for cardiovascular disease.
Lectin-like ox-LDL receptor-1 (LOX-1) with a type II membrane protein structure has been identified as a major endothelial receptor for ox-LDL in endothelial cells (ECs)22). Atherosclerotic stimuli including ox-LDL induce the downregulation of endothelial nitric oxide synthase (eNOS) mediated via LOX-1, which is associated with the activation of small GTP-binding protein RhoA23), and subsequent ox-LDL-reduced nitric oxide (NO) production contributes to impaired endothelial function24,25). MT1-MMP has been shown to be a key effector molecule during NO-induced endothelial migration and tube formation, indicating that MT1-MMP is a potential therapeutic target for NO-associated vascular disorders26). Small GTP-binding protein Rac1, a component of nicotinamide adenine dinucleotide phosphate-oxidase (NADPH oxidase) is activated by ox-LDL, and Rac1 activation subsequently increases reactive oxygen species (ROS) generation, which is implicated in the initiation and progression of atherosclerosis27-29). Molecular links between MT1-MMP and small GTPases—in particular Rho and Rac—have been explored in cell migration as well as molecular synthesis30,31). The formation of a complex of LOX-1 with MT1-MMP was detected by fluorescent immunostaining and immunoprecipitation, and molecular interaction between MT1-MMP and LOX-1 contributes to the RhoA-dependent eNOS protein synthesis and EC invasion, and Rac1-mediated NADPH oxidase activity and ROS generation32) (Fig. 1). The tissue inhibitor of metalloproteinases-2 (TIMP-2) inhibits MT-MMPs, which is an MMP-2 activator, and TIMP-2 binds to the catalytic domain of the cell surface receptor, MT1-MMP in the dimer, and to the hemopexin-like domain of proMMP-233-37). Our findings showed that selective siRNA-mediated suppression of MT1-MMP and TIMP-2 markedly attenuated rapid RhoA and Rac1 activation caused by ox-LDL mediated via LOX-1, which forms a complex with MT1-MMP, confirming that a complex formed with MT1-MMP and LOX-1 plays an integral role in ox-LDL-mediated signaling pathways in ECs32).
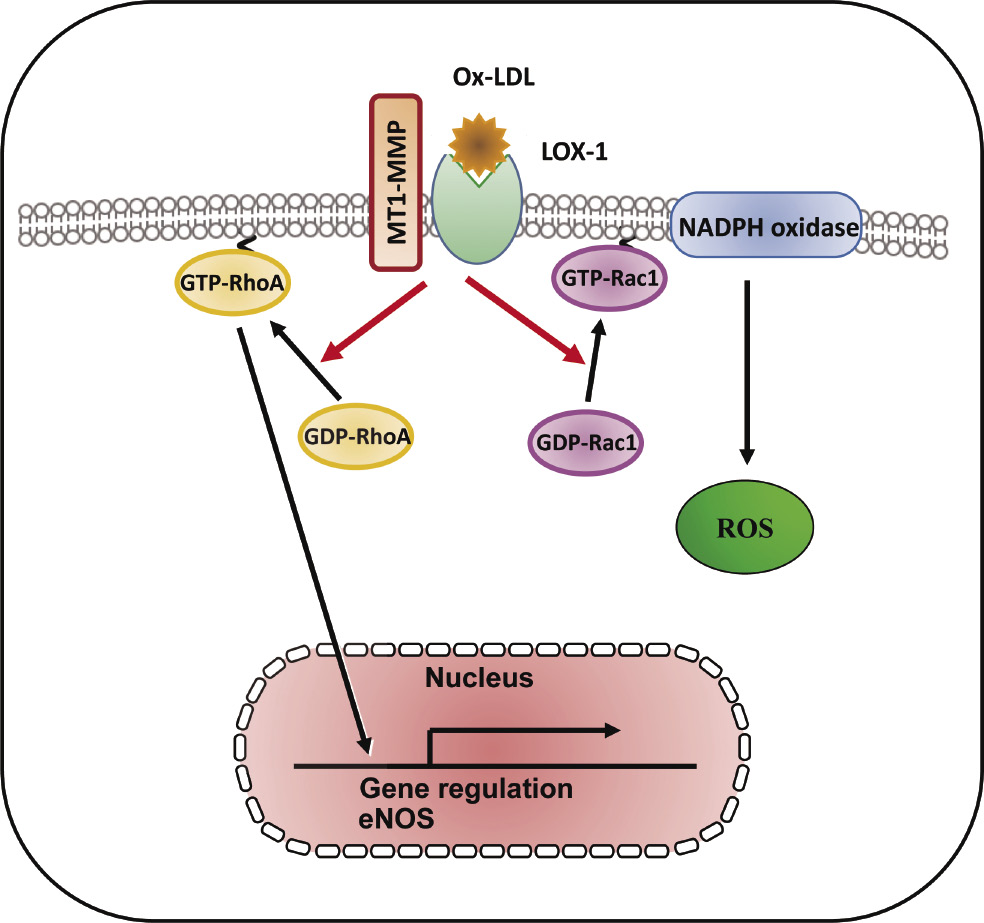
LOX-1-MT1-MMP axis plays a crucial role in RhoA and Rac1 activation signaling pathways in ox-LDL stimulation. In cultured human aortic ECs, surface expression of MT1-MMP is an essential mediator of rapid RhoA and Rac1 activation through the activation of LOX-1 by ox-LDL. The formation of a complex of LOX-1 with MT1-MMP contributed to RhoA-dependent endothelial NO synthase, Rac1-mediated NADPH oxidase activity, and subsequent ROS generation. The LOX-1-MT1-MMP signaling axis plays an important role in ox-LDL-mediated signaling pathways in ECs32).
An advanced glycation end products (AGEs)/receptor for AGE (RAGE) signaling pathways involved in the pathogenesis of microvascular and macrovascular diabetic complications38-40). Ligands which bind to RAGE were accumulated and increased RAGE expression enhanced in accelerated diabetic atherosclerotic lesions41,42). RAGE expression is increased in non-diabetic subjects with premature coronary artery disease43). AGEs induce C-reactive protein expression in hepatoma cells by suppressing Rac1 activation44). AGEs induced Rac1 and p47 (phox), NADPH oxidase activation, resulting in subsequent increased ROS generation and NF-κB phosphorylation-related, redox-sensitive molecular expression in SMCs47) (Fig. 2). Geranylgeranyl transferase I (GGTase I) through the small GTPases RhoA and Rac1 activation is involved in vascular injury with endothelial dysfunction, which contributes to ROS generation and vascular NO production45,46). AGEs induced GGTase I activity, Rac1-p47(phox) activation, NADPH oxidase activity, ROS generation, and molecular expression in SMCs (Fig. 2), and RAGE was found to form a complex with MT1-MMP in both cultured SMCs and the aortae of diabetic rats47). Our findings show the role of MT1-MMP in the AGE/RAGE-triggered signaling pathways and the molecular interaction between RAGE and MT1-MMP in SMCs, suggesting that MT1-MMP may be a novel therapeutic target for diabetic vascular complications47).
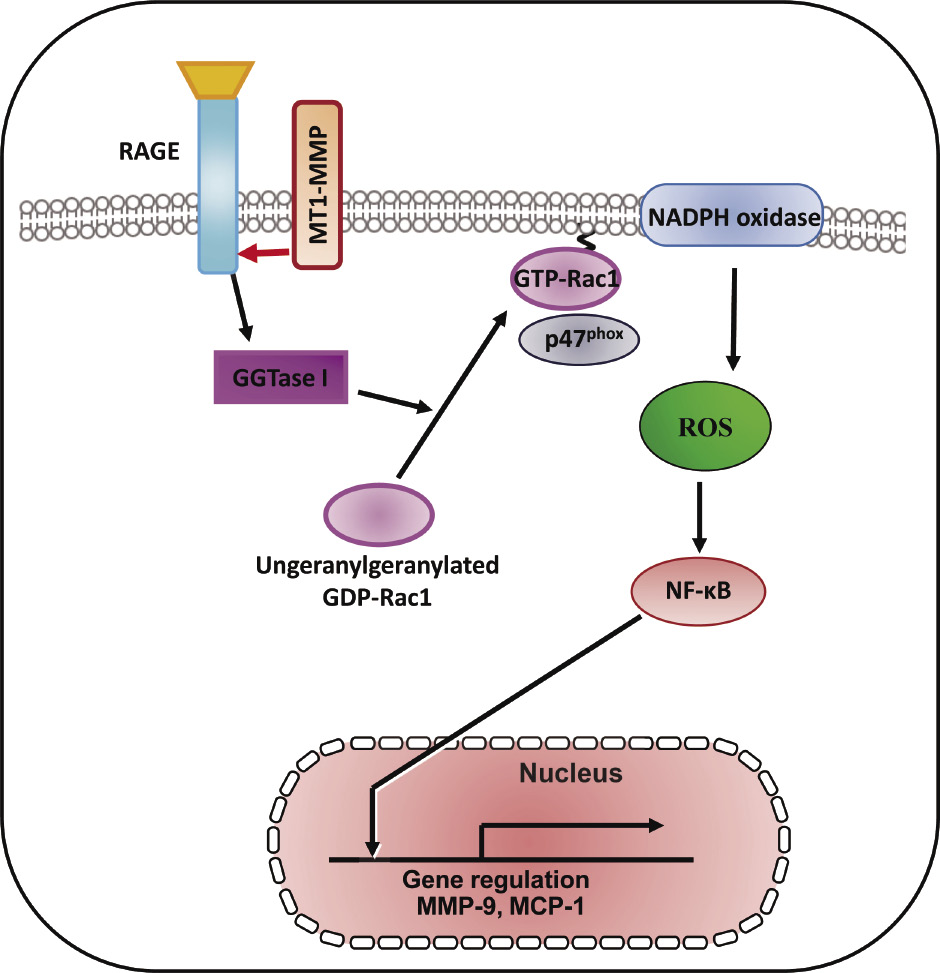
Therapeutic targeting RAGE/MT1-MMP/Rac1 axis in redox-sensitive signaling pathway in diabetic vascular complications. The schematic diagram shows that MT1-MMP is involved in AGEs/RAGE-dependent, redox-sensitive signaling pathways in cultured SMCs. The MT1-MMP/RAGE complex modifies this pathway and the blockade of RAGE/MT1-MMP axis are candidates for therapeutic strategy. These findings identify the attractive therapeutic targeting for RAGE/MT1-MMP/Rac1 in diabetic vascular complications47).
Thrombin regulates the cascade of Rac1/NADPH oxidase-dependent ROS generation, which modulates NO synthesis including eNOS protein expression48,49). Thrombin exerts vascular responses mediated via a family of G protein-coupled receptors, protease-activated receptors (PARs) as well as the process of hemostasis including coagulation, platelet aggregation, and thrombus formation50). Thrombin signaling is mediated by a family of G protein-coupled PARs, and the prototype for this family, PAR1, is activated when thrombin cleaves its N-terminal exodomain at a specific site51). PAR1 is the primary receptor that mediates active responses in atherosclerosis52). Red wine polyphenolic compounds strongly inhibit thrombin-induced matrix invasion of SMCs, and this effect is associated with a direct inhibition of MT1-MMP53). Thrombin upregulates MT1-MMP expression via PI3K and Rac1 activation in cord blood hematopoietic stem/progenitor cells (CB HSPC), and, subsequently, MT1-MMP contributes to the priming of the homing-related responses of CB HSPC54). The interaction between MT1-MMP and PAR1 in the membrane of ECs contributes to the suppression of thrombin-induced endothelial dysfunction via Rac1-dependent NADPH activation including ROS generation and the expression of both TF and the plasminogen activator inhibitor type-1 (PAI-1)60) (Fig. 3A). Silencing of MT1-MMP suppresses sphingosine-1-phosphate-triggered Ca2+ mobilization in glioblastoma cells55). It is known that expression of TF expression is regulated in part by NF-κB phosphorylation56). We previously reported that RhoA-dependent NF-κB phosphorylation and RhoA-related Ca2+ signaling mediate TF and PAI-1 expression in monocyte adhesion to ECs and that there is a cross-talk between Ca2+ signaling and Rac1-dependent ROS generation57,58). Ca2+ influx via transient receptor potential canonical (TRPC) channels induces NF-κB phosphorylation in ECs59). Our study revealed that molecular interaction between MT1-MMP and PAR1 regulated thrombin-triggered TF and PAI-1 overexpression through RhoA-associated Ca2+ signaling and NF-κB phosphorylation in ECs60) (Fig. 3B). Our findings suggest that MT1-MMP mediates thrombin-triggered RhoA and Rac1 activation and Ca2+ signaling in ECs, suggesting that thrombin-triggered MT1-MMP-related signaling may be a target for endothelial dysfunction and oxidant stress in the pathogenesis of cardiovascular diseases60).
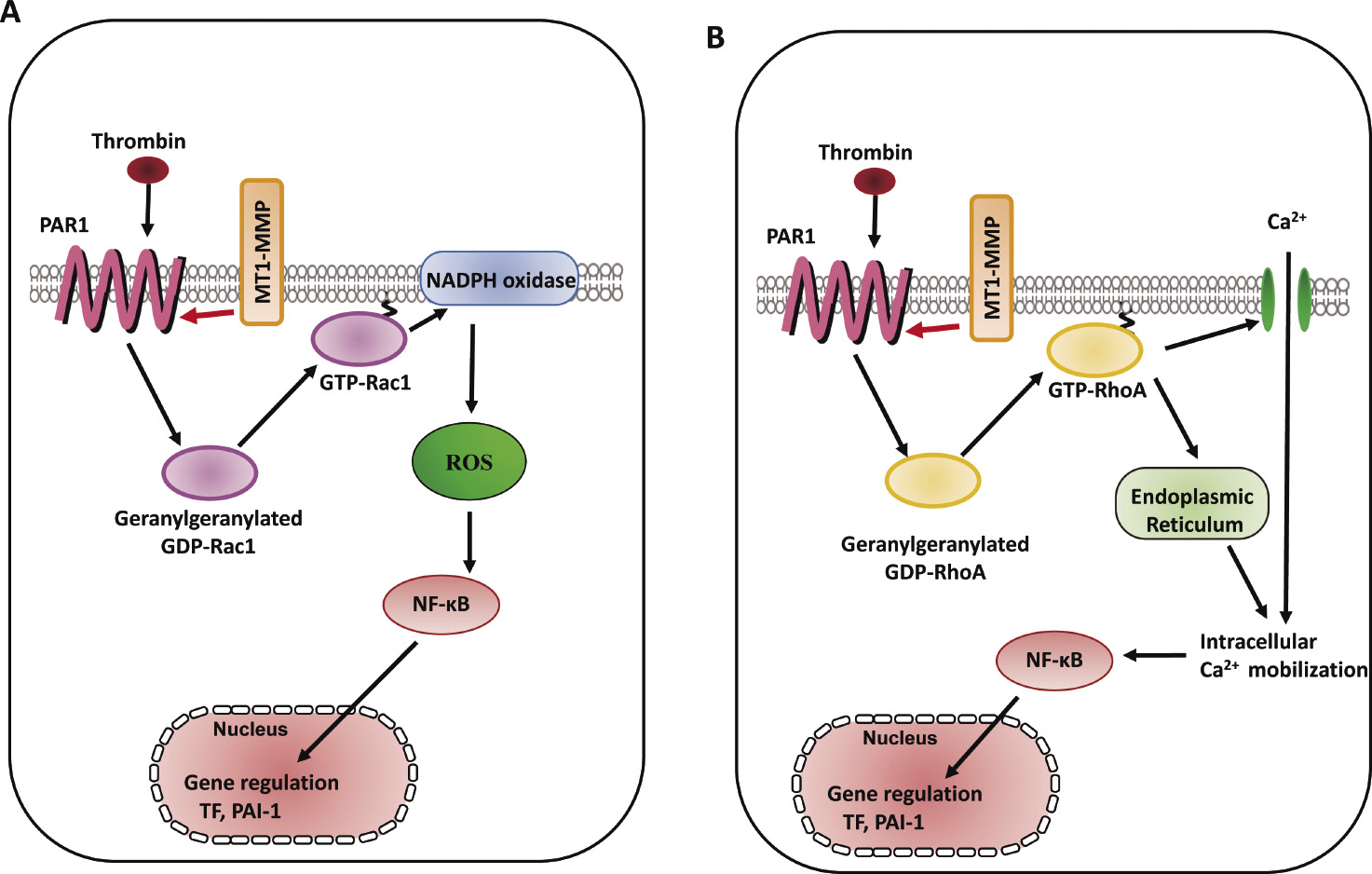
Schema of MT1-MMP involvement in thrombin-triggered signaling pathways in ECs. A. RhoA activation involved in thrombin-triggered [Ca2+]i increase and TF and PAI-1 expression in ECs, whereas Rac1 activation induced thrombin-triggered ROS generation and TF and PAI-1 expression60). B. MT1-MMP mediates thrombin-triggered RhoA and Rac1 activation, resulting in the downstream events including Ca2+ signaling, NADPH oxidase activity, ROS generation, and TF and PAI-1 expression. MT1-MMP contributes to the RhoA/Ca2+ and Rac1/NADPH oxidase-dependent signaling pathways in thrombin-induced vascular responses in ECs60).
Phosphorylation of protein kinase B (Akt) at two key sites, the activation loop and the hydrophobic motif, activates the kinase and promotes endothelial proliferative dysfunction, leading to apoptosis of ECs, and regulates the balance between cell survival61,62). The Akt signaling pathway is also associated with various cellular processes including coagulation and inflammation63). Activation of phospho-inositide 3-kinase (PI3K) and its downstream target Akt is essential for TNF-α-induced NF-κB activation as well as decreased TNF-α-induced adhesion molecule expression and monocyte adhesion, which are linked to the development of vascular diseases and induce inflammatory responses in ECs64). Granulocyte colony-stimulating factor increased MT1-MMP protein and activity in human hematopoietic cells in a PI3K/Akt-dependent manner, indicating the molecular interaction between MT1-MMP and Akt65). Inflammatory cytokines such as TNF-α are master regulators of vascular proatherogenic changes, the action of which has been linked to endothelial dysfunction in many pathophysiological conditions29,66). TNF-α-induced inflammatory responses and procoagulant activity have been implicated in the pathogenesis of vascular diseases67,68). TNF-α induced the increased levels of TF antigen and activity and reduced the antigen levels of thrombomodulin (TM), which directly blocks the interaction between thrombin and the procoagulant protein substrates in the pathogenesis of vascular disease69,70). MT1-MMP possesses transmembrane and cytoplasmic domains in addition to extracellular domains, and the cytoplasmic domain of MT1-MMP has an important role in cell invasion and proliferation, indicating that MT1-MMP functions as a signaling molecule71,72). TIMP-2 and MT1-MMP siRNA enhanced the increased levels of TF antigen and activity, and further reduced the decreased levels of TM antigen in TNF-α-stimulated ECs, indicating that MT1-MMP may be critical for the modulation of procoagulant states in TNF-α stimulation. Additionally, TIMP-2 and MT1-MMP siRNA inhibited the decrease in Akt phosphorylation in TNF-α-stimulated ECs73). A specific pharmacological inhibitor of Akt and Akt siRNA enhanced the TNF-α-induced changes of TF antigen and activity as well as TM antigen in ECs, indicating that MT1-MMP modulates Akt signaling pathways in TNF-α-stimulated ECs73). MT1-MMP siRNA also inhibited TNF-α-induced NF-κB phosphorylation. MT1-MMP binds to Akt within the cytoplasm of TNF-α-treated ECs. These findings suggest that molecular interaction between MT1-MMP and Akt contributes to changes in TNF-α-induced TF and TM expression changes in ECs73) (Fig. 4A). Forkhead box protein O1 (FoxO1) is a transcription factor that contributes to physiological processes including Akt-dependent cell proliferation, apoptosis, and insulin signaling74). TNF-α induced FoxO1 activation via Akt phosphorylation, which acts as a master switch to control cell cycle arrest and apoptosis75,76). Moreover, MT1-MMP regulates EC apoptosis through Akt-mediated phosphorylation of a forehead transcription factor, FoxO1, as well as the activation of caspase-3 in TNF-α stimulation, indicating that the MT1-MMP/Akt signaling axis plays a critical role in the mechanism(s) of endothelial apoptosis73) (Fig. 4B). The MT1-MMP/Akt signaling axis modulates TNF-α-stimulated procoagulant activity and endothelial apoptosis in ECs, suggesting that MT1-MMP is a potential signaling molecule for treating endothelial disordered hemostasis in the development of vascular diseases linked to TNF-α-induced inflammation73).
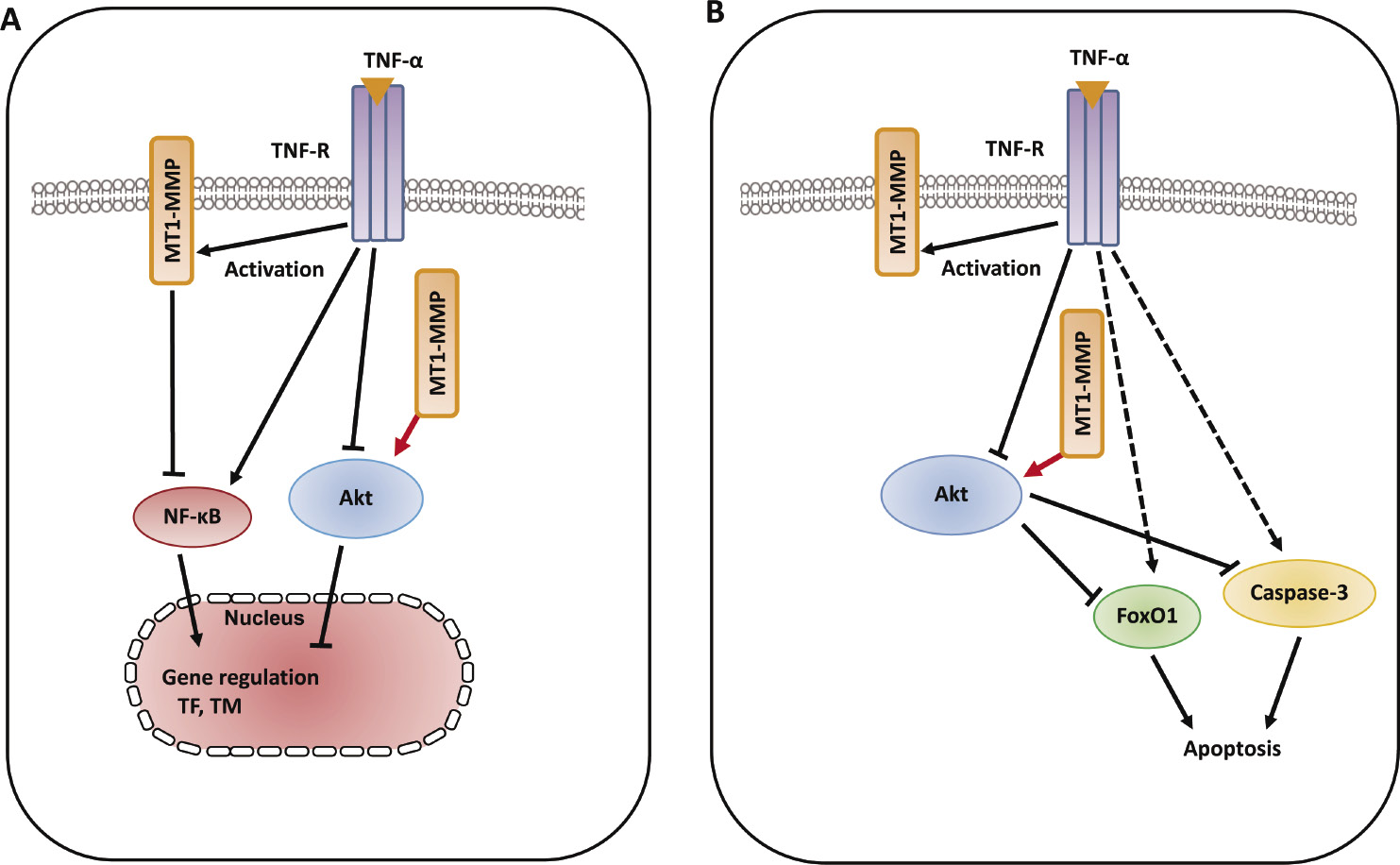
Schematic diagram describing the mechanisms of MT1-MMP/Akt signaling axis in TNF-α-dependent procoagulant activity and apoptosis of ECs. A. MT1-MMP in the cytoplasm of ECs forms a complex with Akt in the intracellular signaling pathways in TNF-α-stimulated ECs. The interaction between MT1-MMP and Akt regulates TNF-α-induced changes in TF and TM expression in ECs73). B. The interaction between MT1-MMP and Akt contributes to endothelial apoptosis through FoxO1 phosphorylation as well as caspase-3 activation. MT1-MMP plays a crucial role of MT1-MMP in Akt-dependent signaling pathways in TNF-α stimulation73).
A proinflammatory cytokine, high-mobility box group 1 (HMGB-1), which is derived from both injured endothelium and activated macrophages/monocytes, contributes to the progression of atherosclerosis and other cardiovascular diseases77). RAGE is a multiligand receptor, which binds to AGEs as well as other high-affinity ligands such as HMGB-178). Statin suppresses vascular inflammation and atherosclerosis in ApoE−/− mice by downregulation of the HMGB-1-RAGE axis in atherosclerotic plaques79). Our previous report has demonstrated that the interaction between MT1-MMP and RAGE induced NADPH oxidase-dependent ROS generation and NF-κB phosphorylation in SMCs47). The binding of MT1-MMP to RAGE also occurred in human ECs80). MMPs are inflammatory mediators linking inflammation with angiogenesis and vascular remodeling81). The addition of HMGB-1 to ECs activated the rapid activation of MT1-MMP80). TIMP-2 and MT1-MMP siRNA prevented the GTP/GDP exchange of small GTP protein RhoA in ox-LDL-stimulated ECs37). The RhoA/Rho kinase pathway was upstream of the NF-κB-dependent pathway, which is required for TF upregulation57,82). Our results clarified that MT1-MMP suppressed HMGB-1-induced TF upregulation via RAGE, RhoA, and NF-κB signaling in ECs80) (Fig. 5). NADPH oxidase in AGE/RAGE-mediated generation of ROS enhanced the expression levels of TF upon stimulation with AGE83). Rac1 activation-dependent ROS production induced thrombin-induced upregulation of TF, which is one of the redox-sensitive, signaling-dependent molecules60). Our findings suggest that HMGB-1-stimulated Rac1 activation induces NADPH oxidase-mediated ROS generation and TF upregulation80) (Fig. 5). Toll-like receptor (TLR)-2 and TLR-4 are expressed in vascular ECs and function as the receptors for HMGB-184). Further studies are needed to examine the role of TLRs in HMGB-1-dependent TF synthesis. Our findings suggest that HMGB-1 activates MT1-MMP through RAGE, leading to RhoA/Rac1 activation and NF-κB phosphorylation, resulting in TF antigen upregulation and eNOS antigen downregulation in the pathogenesis of the progression of endothelial dysfunction-dependent atherosclerosis80).
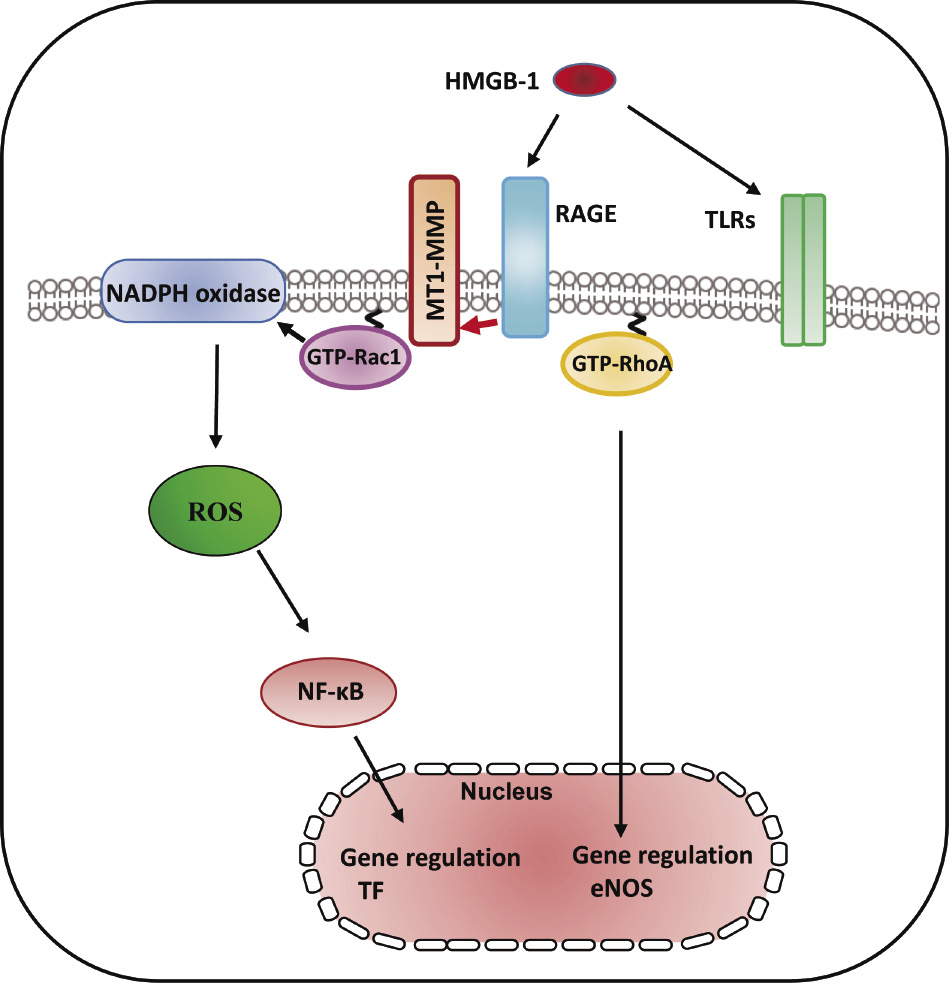
RAGE/MT1-MMP signaling axis modified HMBG-1-mediated TF expression through RhoA and Rac1 activation and NF-κB phosphorylation. HMGB-1 induced RhoA/Rac1 activation and NF-kB phosphorylation in cultured human aortic ECs. HMGB-1 increased the activity of MT1-MMP, which resulted in subsequent activation of RAGE leading to RhoA/Rac1 activation and NF-κB phosphorylation in ECs, indicating that MT1-MMP was involved in vascular inflammation and might be an effective target for treating atherosclerosis80).
In this review, we summarized several features of MT1-MMP as regulators of vascular signaling, which contribute to initiation and progression of atherosclerosis. MT1-MMP is a signaling molecule of vascular proatherogenic changes induced by several proinflammatory and proatherogenic stimuli, which linked to impaired vascular cell function in the pathology of atherosclerosis and subsequent cardiovascular disease32,47,60,73,80).
The authors thank Drs. Koichi Sugimoto, Masashi Kamioka, Atsuya Ando, and Toshiyuki Ishibashi for their contributions to this work.
This work was supported by Fukushima Medical University Research Project and Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (22790718). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.