ABSTRACT
Oxidatively damaged bases in DNA can cause cell death, mutation and/or cancer induction. To overcome such deleterious effects of DNA base oxidation, cells are equipped with base excision repair (BER) initiated by DNA glycosylases. Endonuclease III (Nth), a major DNA glycosylase, mainly excises oxidatively damaged pyrimidines from DNA. The aims of this study were to obtain an overview of the repair mechanism of oxidatively damaged bases and to elucidate the function of BER in maintaining genome stability during embryogenesis and development. In this study, we used the ascidian Ciona intestinalis because at every developmental stage it is possible to observe the phenotype of individuals with DNA damage or mutations. Sequence alignment analysis revealed that the amino acid sequence of Ciona intestinalis Nth homologue (CiNTH) had high homology with those of Escherichia coli, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Caenorhabditis elegans and human Nth homologues. It was evident that two domains, the Helix-hairpin-Helix and 4Fe-4S cluster domains that are critical regions for the Nth activity, are well conserved in CiNTH. CiNTH efficiently complemented the sensitivity of E. coli nth nei mutant to H2O2. CiNTH was bifunctional, with DNA glycosylase and AP lyase activities. It removed thymine glycol, 5-formyluracil and 8-oxoguanine paired with G from DNA via a β-elimination reaction. Interestingly, the N-terminal 44 amino acids were essential for the DNA glycosylase activity of CiNTH.
INTRODUCTION
Reactive oxygen species (ROS) are generated continually in living cells during normal cellular metabolism. ROS are also generated by exogenous sources, such as ionizing radiation and various chemical oxidants. ROS cause various types of oxidative damage to purines and pyrimidines in DNA (Cadet et al., 1997; Wallace, 2002; Bjelland and Seeberg, 2003), which have been implicated in mutation, cancer induction and aging (Ames et al., 1993; Jackson and Loeb, 2001; Tsuzuki et al., 2007). To overcome the deleterious effects of oxidative base damage in DNA, bacteria and eukaryotes have evolved base excision repair (BER) (Dizdaroglu, 2005; David et al., 2007; Hazra et al., 2007; Zharkov, 2008). DNA glycosylases catalyze the first step of the BER pathway by removing modified bases from DNA. DNA glycosylase-associated AP lyase and AP endonuclease recognize the resultant AP site and cleave the phosphodiester backbone at the AP site, followed by DNA repair synthesis by DNA polymerases, and the nick in the DNA backbone is finally rejoined by DNA ligases (Zharkov, 2008; Allinson et al., 2004). Thus, DNA glycosylases are essential to maintain genome stability by excising damaged bases from DNA.
Several DNA glycosylases have been identified that specifically recognize and remove damaged bases from DNA (Dizdaroglu, 2005; Tsuzuki et al., 2007; David et al., 2007; Zharkov, 2008; Hazra et al., 2001). Thymine glycol (Tg), 5-formyluracil (5-foU) and 5-hydroxymethyluracil (5-hmU) are the major oxidative products of thymine (Teebor et al., 1988; Bjelland et al., 2001; Zhang et al., 2000). These lesions are primarily repaired by endonuclease III (Nth) and endonuclease VIII (Nei) in E. coli and by the homologues in S. cerevisiae, S. pombe, C. elegans and human cells (Zhang et al., 1995, 1997, 1999, 2000, 2001, 2003; Bjelland et al., 1994, 1995; Yonekura et al., 2007; Morinaga et al., 2009). The roles of DNA glycosylases in genome stability are also revealed by genetic analyses. E. coli mutants lacking Nth and Nei are highly sensitive to oxidizing agents such as H2O2. E. coli mutants lacking MutM and MutY, which excise damaged purine bases, show about a 1,000-fold increase in the spontaneous mutation frequency (Michaels et al., 1992). DNA glycosylases of many organisms, including E. coli, mice and humans, have been well conserved (Matsumoto et al., 2001; Hazra et al., 2007).
The ascidian (sea squirt) Ciona intestinalis (C. intestinalis) is a multicellular eukaryote that belongs to the subphylum Urochordata, the earliest branch in the phylum Chordata. Nucleotide sequencing of the genome of C. intestinalis has already been achieved (Dehal et al., 2002). Compared to the genome of humans, the ascidian genome is compact (Simmen et al., 1998; Dehal et al., 2002). The haploid genome of C. intestinalis is only about 160 Mbp in size, although it contains about 15,500 genes (Simmen et al., 1998). C. intestinalis is widely used as a model animal to clarify the mechanisms of embryogenesis and regulation of gene expression during development (Satou et al., 2001; Satoh, 2003). It is of interest to clarify how DNA repair mechanisms play a role in the prevention of genome oxidation during development and how these systems have been conserved during evolution. It is also important to characterize the regulation of the expression of BER enzymes during the development of organisms (Vinson and Hales, 2002; Ménézo et al., 2010).
There is only one report on the DNA glycosylases of ascidians. As the first step to address the above questions, it is necessary to identify enzymes that are involved in the repair of oxidatively damaged bases in DNA in C. intestinalis. We recently identified several C. intestinalis homologues of BER enzymes, CiOgg1 (Jin et al., 2006) and AP endonucleases (unpublished results), and the sanitization enzyme for oxidized nucleotides, CiMutT (Yonekura et al., 2010). Knockdown of CiOgg1 expression by a Morpholino oligo (RNAi) method resulted in arrest at an early stage during development in C. intestinalis (Jin et al., 2006). In this study, we first identified an Nth homologue of C. intestinalis (CiNTH). CiNTH robustly complemented the sensitivity of E. coli nth nei mutants to H2O2. Purified CiNTH had Nth activity, i.e. the ability to excise Tg and 5-foU from DNA via a β-elimination reaction. Interestingly, deletion of the N-terminal 44 amino acids abolished the DNA glycosylase activity. CiNTH has a potential role of protecting the genome from oxidative DNA damage caused by ROS in C. intestinalis.
MATERIALS AND METHODS
Chemicals and enzymes
The plasmid vector pGEX-4T-1 and glutathione-Sepharose 4B were obtained from Amersham Pharmacia Biotech (Uppsala, Sweden). KOD (Thermococcus kodakaraensis) DNA polymerase was purchased from Toyobo (Osaka, Japan). [γ-32P]ATP (>259 TBq/mmol) was obtained from MP Biomedicals (Costa Mesa, CA). Other high performance liquid chromatography-purified oligonucleotides were obtained from Takara Shuzo (Kyoto, Japan). Protease inhibitor cocktail and isopropyl-1-thio-β-D-galactopyranoside (IPTG) were obtained from Nacalai Tesque (Kyoto, Japan).
Identification and cloning of Nth homologue of C. intestinalis
The Ghost database was searched for proteins with homology to E. coli Nth (EcNth) and human NTH1 using the BLAST database. The cima848a06 clones were thereby detected. The Ciona cima848a06 cDNA clone was amplified by PCR with KOD-Plus DNA polymerase. The PCR primers used were: for forward with a BamHI site 5’-CATGGATCCATGCATCGTTACGTTCA-3’ and for reverse with a XhoI site 5’-CGCCTCGAGAATTACTAAGACACAATTTATTT-3’. The pGEX-4T-1 plasmid was used as cloning and expression vector. The PCR products were cloned into the pGEX-4T-1 vector digested with appropriate restriction enzymes, and then E. coli SY5 nth nei was transformed with the plasmid pGEX-CiNTH. The sequence of the insert was checked to verify that no mutations had been introduced by the PCR.
Expression and purification of CiNTH
A single colony of E. coli SY5 nth nei mutant carrying pGEX-CiNTH was incubated in 30 ml of LB containing 100 μg/ml of ampicillin and grown overnight at 37°C. Thirty milliliters of the overnight culture were added to 3 liters of fresh LB (1:100 dilution) containing 100 μg/ml of ampicillin and grown at 37°C until the optical density at 600 nm reached about 0.6. The culture was further incubated overnight at 15°C in the presence of 0.01 mM IPTG. The induced cells were harvested, washed and resuspended in buffer A (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 2 mM EDTA containing 0.02% Triton X-100). The cell suspension was sonicated and the cell lysate was centrifuged at 20,000 g at 4°C for 30 min. The supernatant was applied to a glutathione (GSH)–Sepharose 4B column (Pharmacia Biotech) that had been equilibrated with buffer A. The purified GST-CiNTH fusion protein was eluted from the column with buffer B (50 mM Tris-HCl, pH 9.6, 40 mM GSH, 30 mM NaCl), followed by dialysis overnight at 4°C against buffer C [100 mM Tris-HCl, pH 8.0, 60 mM NaCl, 2 mM EDTA, 0.04% TritonX-100 and 25% glycerol, 13.4 mM 2-mercaptoethanol (EtSH)], and stored at –80°C until use. Proteins were analyzed by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE). Protein concentration was determined by the Bradford method with bovine serum albumin (BSA) as a standard.
DNA substrates
Tg-containing oligonucleotide was prepared as previously described (Dianov et al., 2000). Briefly, oligonucleotide containing a single thymine residue was oxidized with 50 mM osmium tetroxide in the presence of 2% pyridine at room temperature for 15 min, followed by purification by gel filtration on Sephadex G-25. 5-foU-containing oligonucleotide was synthesized by sodium periodate oxidation of 5-(1,2-dihydroxyethyl)uracil-containing oligonucleotide (Sugiyama et al., 1996). The 8-oxoG-containing oligonucleotide was obtained from Trevigen (Gaithersburg, MD). The oligonucleotides containing Tg, 5-foU and 8-oxoG were labeled at the 5'-end by [γ-32P]ATP using T4 polynucleotide kinase (Toyobo) and annealed with complementary strands as previously described (Zhang et al., 1997, 1998). The nucleotide sequences of the oligonucleotides are shown in Fig. 1.
DNA cleavage assay
20 fmol of 32P-labeled duplex oligonucleotide was incubated with 5 pmol of GST-CiNTH or 500 fmol of GST-EcNth in a reaction mixture (10 μl) containing 50 mM Tris-HCl, pH 6.8, 1 mM EDTA, 1 mM DTT, 0.1 mg/ml BSA and 75 mM NaCl. The reaction was carried out at 26°C for 30 min or other conditions as indicated. The reaction was terminated by the addition of stop solution (95% formamide, 0.1% bromophenol blue, 0.1% xylene cyanol and 20 mM EDTA). The samples were then heated at 95oC for 5 min, immediately cooled on ice, and loaded onto 20% polyacrylamide gels in Tris-borate, pH 8.3 containing 7 M urea and 2 mM EDTA. After electrophoresis at 1300 V for 90 min, the gels were autoradiographed using BAS-1800 II (Fuji Film, Tokyo). Quantitative analysis of the band intensity was performed using an ImageJ software.
Trapping assay
20 fmol of 32P-labeled duplex oligonucleotide was incubated with 5 pmol of GST-CiNTH or 5 pmol of GST-EcNth in a reaction mixture (15 μl) containing 50 mM Tris-HCl, pH 8.0, 1 mM EDTA in the presence of 100 mM NaBH4. The reaction was carried out at 26 or 37°C for 60 min and terminated by heating at 95°C for 5 min after the addition of 2 x standard sample buffer for SDS-PAGE. Trapped protein-oligonucleotide complexes were separated by 12% SDS-PAGE and the gels were autoradiographed using BAS-1800 II.
Complementation assay
Overnight cultures of E. coli SY5, SY5 nth nei with pGEX-4T-1 vector and SY5 nth nei containing pGEX-CiNTH were appropriately diluted and plated on LB agar containing H2O2 (up to 450 μM). After incubation at 37°C for 42 hr, the number of colonies was counted to estimate survival.
Construction of N-terminal-truncated CiNTH
To prepare an expression vector of C-terminally GST-fused protein, first, a GST cDNA fragment containing a stop codon was PCR-amplified from the pGEX-4T-1 vector. The amplified fragment containing XhoI and SalI sites at its 5’ and 3’ ends, respectively, was inserted into an XhoI site in pET21a (+) vector (Novagen). The obtained vector was designated pET-GST vector. Full-length (FL) and four N-terminal deletion fragments, dN42, dN43, dN44 and dN45, were PCR-amplified from the pGEX-CiNTH expression vector, and inserted into NdeI and XhoI sites in pET-GST vector. Purification of FL and N-terminal-truncated CiNTH-GST fusion proteins was performed as described above with the exception of using BL21(DE3) strain as a host strain.
RESULTS
Identification of C. intestinalis homologues of human NTH1
A search for candidates for the ascidian C. intestinalis homologues of human NTH1 was done by using the NCBI-BLAST database using the full-length amino acid sequence of human NTH1. Among several candidates, one cDNA clone, cima848a06, coding for a peptide with a high degree of homology to human and mouse NTH1 was identified in the EST database from the Ciona cDNA resources. The cDNA of CiNTH is 795 bp long and codes for a 265 amino acid polypeptide. Sequence alignments showed that the encoded protein, CiNTH, shares 48%, 40% and 23% identity of the amino acid sequence with the Homo sapiens, C. elegans and E. coli Nth homologues, respectively (Fig. 2). CiNTH contained well-conserved lysine (Lys176) and aspartic acid (Asp195) residues that have counterparts in human NTH1 (Lys220 and Asp239) (Ikeda et al., 1998). The polypeptide had two well-known domains, the Helix-hairpin-Helix (HhH) and iron-sulfur binding domains (Denver et al., 2003; Lukianova and David, 2005). The HhH domain is a well-conserved DNA-binding domain found in several types of DNA glycosylases, including Nth, Ogg1, MutY and AlkA (Denver et al., 2003). The conserved HhH structural elements are labeled C in Fig. 2. The 4Fe-4S motif (labeled box E), which plays a major role in establishing the high degree of opposite-strand specificity displayed by NTH1 enzymes, was also highly conserved (Golinelli et al., 1999; Liu et al., 2003; Porello et al., 1998; Roldán-Arjona et al., 1996). CiNTH has a mitochondrial localization signal (MLS) located at the N-terminus, according to the prediction by the PSORT II algorithm (Fig. 2).

To determine whether CiNTH is required for the cellular response to DNA damage in vivo, we transformed and expressed GST-CiNTH in E. coli SY5 nth nei and compared the sensitivity to H2O2 with those of wild-type SY5 and SY5 nth nei bearing the vector alone (Fig. 3). E. coli SY5 nth nei mutant was more sensitive to H2O2 than the wild-type SY5. This is due to the mutant’s lack of the full ability to repair oxidized pyrimidine bases generated by H2O2. Expression of GST-CiNTH in E. coli SY5 nth nei partially complemented the sensitivity to H2O2, like that of E. coli Nth, while the mutant carrying vector pGEX-4T-1 was highly sensitive to the agent. These results indicated that CiNTH could function as a DNA glycosylase to repair damaged pyrimidine bases.
Expression and purification of CiNTH protein
To clarify whether CiNTH has DNA glycosylase activity, GST-CiNTH fusion protein was expressed in E. coli SY5 nth nei. The nth nei mutant cells were used to avoid contamination with E. coli Nth and Nei during purification of CiNTH protein. The GST-CiNTH fusion protein and E. coli GST-Nth were induced with IPTG and purified by GSH-Sepharose column chromatography (Fig. 4A). Purified GST-CiNTH gave a major band with an apparent molecular mass of ~56 kDa on 12% SDS-PAGE. EcNth and CiAPE (C. intestinalis AP endonuclease) were also purified (Fig. 4A).
Cleavage activity of CiNTH on Tg-containing duplex oligonucleotide
DNA cleavage assays were carried out using the purified GST-CiNTH and GST-EcNth with Tg-containing 17-mer duplex oligonucleotide as a substrate. The results are shown in Fig. 4, B and C. CiNTH could cleave the Tg-containing oligonucleotide at the Tg site by β-elimination, like EcNth. Further reaction of the products with CiAPE generated a 3’-OH on the CiNTH product (Fig. 4C). These results indicated that CiNTH has the same type of endonuclease III activity as its E. coli, C. elegans and human counterparts (Ikeda et al., 1998; Miyabe et al., 2002; Morinaga et al., 2009).
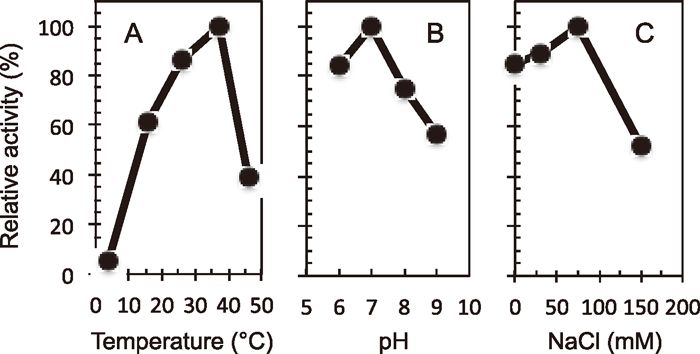
The activity of CiNTH at various temperatures, pHs and NaCl concentrations
To characterize the properties of CiNTH, we carried out the cleavage assay at different temperatures (Fig. 5A). The results showed that the optimal temperature of CiNTH enzymatic activity was 37°C, not 26°C, which is the optimal temperature for CiOgg1 DNA glycosylase (Jin et al., 2006).
The optimal pH of the DNA glycosylase activity of CiNTH was determined at 37°C in reaction mixtures at pH 6.0–9.0. As shown in Fig. 5B, CiNTH had a pH optimum of 7.0, which was similar to those of EcNth and human NTH1 (Ikeda et al., 1998). The DNA glycosylase activity of CiNTH at various concentrations of NaCl was also determined. The results are shown in Fig. 5C. The optimal concentration of NaCl for the CiNTH activity was around 75 mM.
DNA glycosylase/AP lyase activity of CiNTH on various substrates
Substrate oligonucleotide (20 fmol) containing Tg, 5-foU or 8-oxoG paired with G, A, T or C was incubated at 37°C for 5 min with 5 pmol of purified GST-CiNTH. The results are shown in Fig. 6A. CiNTH cleaved the oligonucleotides at the sites of Tg or 5-foU paired with any of the four bases via a β-elimination reaction to generate a 9-mer product. In contrast, it excised 8-oxoG when paired with G, and weak but detectable activity could also be seen with base pairs other than 8-oxoG:G. This substrate specificity was similar to those of E. coli, S. pombe and human NTH proteins (Matsumoto et al., 2001; Yonekura et al., 2007).

The trapping assay is based on the fact that DNA glycosylase with AP lyase activity forms Schiff base intermediates that can be trapped by reducing agents such as NaBH4 to generate stable enzyme-DNA complexes (Rabow and Kow, 1997; Zhang et al., 2000). The trapping assay was carried out with purified GST-CiNTH to examine whether CiNTH actually had AP lyase activity. Oligonucleotides containing Tg, 5-foU or 8-oxoG paired with G, A, T or C were incubated with purified GST-CiNTH in the presence of 100 mM NaBH4. The trapped protein-DNA complexes were analysed by 12% SDS-PAGE. As shown in Fig. 6B, CiNTH was trapped by the duplex oligonucleotides containing Tg or 5-foU paired with G, A, T or C. CiNTH was also trapped by the oligonucleotide containing 8-oxoG paired with G, A, T or C.
The N-terminal amino acid sequence has critical function for CiNTH activity
As shown in Fig. 7A, although the N-terminal extensions in eukaryotic Nth homologues have high diversity, the N-terminal regions of human NTH1 and C. elegans CeNTH play an essential role in the endonuclease III activity (Liu and Roy, 2002; Morinaga et al., 2009). To examine whether the N-terminal amino acids influence the CiNTH activity, we performed N-terminal deletion experiments. We examined the effect of deletion of the amino acid sequence in CiNTH on its DNA glycosylase/AP lyase activity. For these experiments, GST tag was fused to the C-terminus of CiNTH fragments (Fig. 7B). The deletion plasmids were constructed by PCR, and fragments were sub-cloned into pET-GST plasmid. E. coli BL21 (DE3) was transformed with the plasmids and the fusion proteins were overexpressed by adding IPTG. The N-terminal truncated CiNTH-GST proteins were partially purified using GSH-Sepharose beads (Fig. 7C). DNA glycosylase/AP lyase activity was assayed by trapping with Tg, 5-foU, or 8-oxoG-containing duplex oligonucleotide. The results showed that deletion of the N-terminal 45 amino acids completely abolished the CiNTH activity on duplex oligonucleotide containing Tg (Fig. 7D), 5-foU (Fig. 7E) or 8-oxoG (Fig. 7F). On the other hand, deletion of 44 residues from the N-terminus did not affect the catalytic activity (Fig. 7, D–F). These results demonstrated that the amino acid residue located 45 residues from the N-terminus plays a critical role for the endonuclease III activity of CiNTH.

DISCUSSION
ROS induce oxidative damage to DNA, including various types of oxidative modifications to purine and pyrimidine bases, which might be involved in a wide array of biological processes, including mutagenesis, cancer induction and aging (Martin et al., 1996; Demple and Harrison, 1994; Ames and Gold, 1991). BER is an essential cellular system that suppresses the deleterious effects of damaged bases in DNA. Endonuclease III (Nth) is the main DNA glycosylase that excises oxidatively damaged pyrimidine bases such as Tg and 5-foU as the first step of BER. The biological importance of Nth is supported by the strong conservation of its amino acid sequence from prokaryotes to humans.
Little is known about how DNA damage processing is related to development and early stages of growth. C. intestinalis is a good model system not only for studying development-related biological phenomena (Satoh, 1994, 2003; Simmen et al., 1998; Sasakura et al., 2003; Yagi et al., 2004) but also for investigating developmental and tissue-specific DNA repair mechanisms. These considerations led us to examine how the BER system for oxidatively damaged bases is conserved in the ascidian C. intestinalis. In previous studies we observed that CiOgg1 is highly expressed in the cleaving embryo stage during the development of C. intestinalis (Jin et al., 2006). Interestingly, EST analysis using the Ghost Database (Ciona intestinalis Genome and cDNA Resources in Japan, Kyoto University) showed that CiNTH is highly expressed in the endostyle and heart. It is necessary to know how the expression of CiOgg1 and CiNTH is regulated during development and how the regulation of these enzymes influences the development of C. intestinalis.
The amino acid sequences of Nth family proteins exhibit highly conserved regions in their primary structure, namely, HhH and 4Fe-4S cluster domains (Fig. 2). CiNTH has significant homology with EcNth and human NTH1, including five DNA-binding motifs and catalytic residues Lys and Asp (Ikeda et al., 1998). However, the N-terminal region (residues 1–45) of CiNTH is not present in EcNth (Figs. 2 and 7A). Previously we showed that the N-terminal region of CeNTH is required for its DNA glycosylase/AP lyase activity (Morinaga et al., 2009). This study revealed that the N-terminal region of CiNTH also plays a critical role for the DNA glycosylase/AP lyase activity. Human NTH1 also has an N-terminal tail (residues 1–95) that is essential for the enzymatic activity (Liu and Roy, 2002). However, the amino acid sequences of these N-terminal tails do not share any homology in human, C. elegans and C. intestinalis.
We searched the BLAST database using the amino acid sequence positioned at 1–44 from CiNTH as a query, but no protein with significant similarity was identified. There are no known motifs in this region. The homology to the N-terminal region of human NTH1 and CeNTH was very low. Studies to clarify the significance of the N-terminal amino acids are now underway in our laboratory.
We examined the effect of deletion of the N-terminal amino acid sequence in CiNTH on its DNA glycosylase activity. Deletion of 44 residues from the N-terminus did not affect its catalytic activity (Fig. 7, D–F). In contrast, deletion of 45 residues from the N-terminus completely abolished the trapping activity against duplex oligonucleotides containing Tg (Fig. 7D), 5-foU (Fig. 7E) or 8-oxoG (Fig. 7F). A previous report revealed that the wild-type and 1-80-truncated human NTH1 protein had normal NTH activity, while the 1-92-truncated human NTH1 did not (Liu and Roy, 2002). In our preliminary data, the dN47 CiNTH fragment fused to GST at the N-terminus had Tg-DNA glycosylase activity, and this activity was still remained in the dN47 CiNTH fragment after removal of the GST-tag by thrombin protease (data not shown). In this fragment, however, five amino acids, Gly-Ser-Pro-Gln-Phe, still remained at positions 43–47, even after digestion with thrombin. The amino acid residues at positions 43–47 in CiNTH is Tyr-Glu-Pro-Glu-His. Together with the data shown in Fig. 7 and our preliminary data, this suggests that the proline residue at position 45 in CiNTH might be important for Nth activity. Supporting this idea, this proline residue is located just before the first methionine residue of EcNth, and is highly conserved in H. sapiens, Mus musculus, C. intestinalis, S. pombe (Fig. 7A), suggesting that this proline residue has roles in the N-terminal capping of NTH catalytic polypeptides and in forming the bridge between the catalytic core and the N-terminal extension, which is evolved in eukaryotes. Sakai and co-workers prepared a series of C-terminal truncated human MTH1 fragments, and investigated their stability and enzymatic activity (Sakai et al., 2002). The results suggested that the C-terminal region is essential for maintaining a stable conformation of human MTH1 protein. It should be noted that our deletion fragments had different stabilities (data not shown). FL-GST, dN42-GST and dN43-GST could be produced with good yield, whereas the expression levels of fragments dN44-GST and dN45-GST were extremely reduced. In the case of dN46-GST, further reduction of the expression level in E. coli was observed. These observations also support the idea that the amino acid residues around the proline may contribute to the stability of CiNTH.
We previously observed that knockdown of CiOgg1 expression by a Morpholino oligo (RNAi) method resulted in an arrest of the early stage during development in C. intestinalis (Jin et al., 2006). Higher oxygen consumption may correlate with a higher level of oxidative stress, and the level of expression of the CiOgg1 and CiNTH genes must be regulated to respond to such higher oxidative stress conditions. Experiments to clarify the roles of these proteins in development are currently in progress.
ACKNOWLEDGMENT
We thank Drs. Yutaka Satou and Noriyuki Satoh for kindly supplying Ciona cDNA plasmids. We also thank Drs. Elizabeth Nakajima and Shuji Yonei for critically reading the manuscript. This research was financially supported in part by the Global Center of Excellence Program “Formation of a Strategic Base for Biodiversity and Evolutionary Research (A06): from Genome to Ecosystem” and Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports and Technology of Japan.
REFERENCES
- Allinson, S. L., Sleeth, K. M., Matthewman, G. E., and Dianov, G. L. (2004) Orchestration of base excision repair by controlling the rates of enzymatic activities. DNA Repair 3, 23–31.
- Ames, B. N., and Gold, L. S. (1991) Endogenous mutagens and the causes of aging and cancer. Mutat. Res. 250, 3–16.
- Ames, B. N., Shigenaga, M. K., and Hagen, T. M. (1993) Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 90, 7915–7922.
- Bjelland, S., and Seeberg, E. (2003) Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat. Res. 531, 37–80.
- Bjelland, S., Birkeland, N. K., Benneche, T., Volden, G., and Seeberg, E. (1994) DNA glycosylase activities for thymine residues oxidized in the methyl group are functions of the AlkA enzyme in Escherichia coli. J. Biol. Chem. 269, 30489–30495.
- Bjelland, S., Eide, L., Time, R. W., Store, R., Eftedal, I., Volden, G., and Seeberg, E. (1995) Oxidation of thymine to 5-formyluracil in DNA: mechanisms of formation, structural implications, and base excision by human cell free extracts. Biochemistry 34, 14758–14764.
- Bjelland, S., Ånensen, H., Knævelsrud, I., and Seeberg, E. (2001) Cellular effects of 5-formyluracil in DNA. Mutat. Res. 486, 147–154.
- Cadet, J., Berger, M., Douki, T., and Ravanat, J. L. (1997) Oxidative damage to DNA: formation, measurement, and biological significance. Rev. Physiol. Biochem. Pharmacol. 131, 1–87.
- David, S. S., O’Shea, V. L., and Kundu, S. (2007) Base-excision repair of oxidative DNA damage. Nature 447, 941–950.
- Dehal, P., Satou, Y., Campbell, R. K., Chapman, J., Degnan, B., De Tomaso, A., Davidson, B., Di Gregorio, A., Gelpke, M., et al. (2002) The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science 298, 2157–2167
- Demple, B., and Harrison, L. (1994) Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 63, 915–948.
- Denver, D. R., Swenson, S. L., and Lynch, M. (2003) An evolutionary analysis of the helix–hairpin–helix superfamily of DNA repair glycosylases. Mol. Biol. Evol. 20, 1603–1611.
- Dianov, G. L., Thybo, T., Dianova, I. I., Lipinski, L. J., and Bohr, V. A. (2000) Single nucleotide patch base excision repair is the major pathway for removal of thymine glycol from DNA in human cell extracts. J. Biol. Chem. 275, 11809–11813.
- Dizdaroglu, M. (2005) Base-excision repair of oxidative DNA damage by DNA glycosylases. Mutat. Res. 591, 45–59.
- Golinelli, M. P., Chmiel, N. H., and David, S. S. (1999) Site-directed mutagenesis of the cysteine ligands to the [4Fe–4S] cluster of Escherichia coli MutY. Biochemistry 38, 6997–7007.
- Hazra, T. K., Hill, J. W., Izumi, T., and Mitra, S. (2001) Multiple DNA glycosylases for repair of 8-oxoguanine and their potential in vivo functions. Prog. Nucleic Acids Res. Mol. Biol. 68, 193–205.
- Hazra, T. K., Das, A., Das, S., Choudhury, S., Kow, Y. W., and Roy, R. (2007) Oxidative DNA damage repair in mammalian cells: a new perspective. DNA Repair 6, 470–480.
- Ikeda, S., Biswas, T., Roy, R., Izumi, T., Boldogh, I., Kurosky, A., Sarker, A. H., Seki, S., and Mitra, S. (1998) Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. J. Biol. Chem. 273, 21585–21593.
- Jackson, A. L., and Loeb, L. A. (2001) The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat. Res. 477, 7–21.
- Jin, G., Zhang, Q.-M., Satou, Y., Satoh, N., Kasai, H., and Yonei, S. (2006) Cloning and characterization of an ascidian homolog of the human 8-oxoguanine DNA glycosylase (Ogg1) that is involved in the repair of 8-oxo-7, 8-dihydroguanine in DNA in Ciona intestinalis. Int. J. Radiat. Biol. 82, 241–250.
- Liu, X., and Roy, R. (2002) Truncation of amino-terminal tail stimulates activity of human endonuclease III (hNTH1). J. Mol. Biol. 321, 265–276.
- Liu, X., Choudhury, S., and Roy, R. (2003) In vitro and in vivo dimerization of human endonuclease III stimulates its activity. J. Biol. Chem. 278, 50061–50069.
- Lukianova, O. A., and David, S. S. (2005) A role for iron–sulfur clusters in DNA repair. Curr. Opin. Chem. Biol. 9, 145–151.
- Martin, G. M., Austad, S. N., and Johnson, T. E. (1996) Genetic analysis of ageing: role of oxidative damage and environmental stresses. Nat. Genet. 13, 25–34.
- Matsumoto, Y., Zhang, Q.-M., Takao, M., Yasui, A., and Yonei, S. (2001) Escherichia coli Nth and human hNTH1 DNA glycosylases are involved in removal of 8-oxoguanine from 8-oxoguanine/guanine mispairs in DNA. Nucleic Acids Res. 29, 1975–1981.
- Ménézo, Y., Dale, B., and Cohen, M. (2010) DNA damage and repair in human oocytes and embryos: a review. Zygote 21, 1–9.
- Michaels, M. L., Cruz, C., Grollman, A. P., and Miller, J. H. (1992) Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl. Acad. Sci. USA 89, 7022–7025.
- Miyabe, I., Zhang, Q.-M., Kino, K., Sugiyama, H., Takao, M., Yasui, A., and Yonei, S. (2002) Identification of 5-formyluracil DNA glycosylase activity of human hNTH1 protein. Nucleic Acids Res. 30, 3443–3448.
- Morinaga, H., Yonekura, S., Nakamura, N., Sugiyama, H., Yonei, S., and Zhang-Akiyama, Q.-M. (2009) Purification and characterization of Caenorhabditis elegans NTH, a homolog of human endonuclease III: Essential role of N-terminal region. DNA repair 8, 844–851.
- Porello, S. L., Cannon, M. J., and David, S. S. (1998) A substrate recognition role for the [4Fe–4S]2+ cluster of the DNA repair glycosylase MutY. Biochemistry 37, 6465–6475.
- Rabow, L. E., and Kow, Y. W. (1997) Mechanism of action of base release by Escherichia coli Fpg protein: role of lysine 155 in catalysis. Biochemistry 36, 5084–5096.
- Roldán-Arjona, T., Anselmino, C., and Lindahl, T. (1996) Molecular cloning and functional analysis of a Schizosaccharomyces pombe homologue of Escherichia coli endonuclease III. Nucleic Acids Res. 24, 3307–3312.
- Sakai, Y., Furuichi, M., Takahashi, M., Mishima, M., Iwai, S., Shirakawa, M., and Nakabeppu, Y. (2002) A molecular basis for the selective recognition of 2-hydroxy-dATP and 8-oxo-dGTP by human MTH1. J. Biol. Chem. 277, 8579–8587.
- Sasakura, Y., Shoguchi, E., Takatori, N., Wada, S., Rokhsar, D., Levine, M., Kohara, Y., Satou, Y., and Satoh, N. (2003) A genomewide survey of developmentally relevant genes in Ciona intestinalis. X. Genes for cell junctions and extracellular matrix. Dev. Genes Evol. 213, 303–313.
- Satoh, N. (1994) Developmental Biology of Ascidians. Cambridge University Press, New York.
- Satoh, N. (2003) Ascidian embryos as a model system to analyze expression and function of development genes. Differentiation 68, 1–12.
- Satou, Y., Takatori, N., Yamada, L., Mochizuki, Y., Hamaguchi, M., Ishikawa, H., Chiba, S., Imai, K., Kano, S., et al. (2001) Gene expression profiles in Ciona intestinalis tailbud embryos. Development 128, 2893–2904.
- Simmen, M., Leitgeb, S., Clark, V. H., Jones, S. J., and Bird, A. (1998) Gene number in an invertebrate chordate, Ciona intestinalis. Proc. Natl. Acad. Sci. USA 95, 4437–4440.
- Sugiyama, H., Matsuda, S., Zhang, Q.-M., Yonei, S., and Saito, I. (1996) New synthetic method of 5-formyluracil-containing oligonucleotides and their melting behavior. Tetrahedron Lett. 37, 9067–9070.
- Teebor, G. W., Boorstein, R. J., and Cadet, J. (1988) The repairability of oxidative free radical mediated damage to DNA: a review. Int. J. Radiat. Biol. 54, 131–150.
- Tsuzuki, T., Nakatsu, Y., and Nakabeppu, Y. (2007) Significance of error-avoiding mechanisms for oxidative DNA damage in carcinogenesis. Cancer Sci. 98, 465–470.
- Vinson, R. K., and Hales, B. F. (2002) DNA repair during organogenesis. Mutat. Res. 509, 79–91.
- Wallace, S. S. (2002) Biological consequences of free radical-damaged DNA bases. Free Radical Biol. Med. 33, 1–14.
- Yagi, K., Satoh, N., and Satou, Y. (2004) Identification of downstream genes of the ascidian muscle determinant gene Ci-macho1. Dev. Biol. 15, 478–489.
- Yonekura, S., Nakamura, N., Doi, T., Sugiyama, H., Yamamoto, K., Yonei, S., and Zhang, Q.-M. (2007) Recombinant Schizosaccharomyces pombe Nth1 protein exhibits DNA glycosylase activities for 8-oxo-7,8-dihydroguanine and thymine residues oxidized in the methyl group. J. Radiat. Res. 48, 417–424.
- Yonekura, S., Sanada, U, and Zhang, Q.-M. (2010) CiMutT, an asidian MutT homologue, has a 7, 8-dihydro-8-oxo-dGTP pyrophosphohydrolase activity responsible for sanitization of oxidized nucleotides in Ciona intestinalis. Genes Genet. Syst. 85, 287–295.
- Zhang, Q.-M. (2001) Role of the Escherichia coli and human DNA glycosylases that remove 5-formyluracil from DNA in the prevention of mutations. J. Radiat. Res. 42, 11–19.
- Zhang, Q.-M., Fujimoto, J., and Yonei, S. (1995) Enzymatic release of 5-formyluracil by mammalian liver extracts from DNA irradiated with ionizing radiation. Int. J. Radiat. Biol. 68, 603–607.
- Zhang, Q.-M., Sugiyama, H., Miyabe, I., Matsuda, S., Saito, I., and Yonei, S. (1997) Replication of DNA templates containing 5-formyluracil, a major oxidative lesion of thymine in DNA. Nucleic Acids Res. 25, 3969–3973.
- Zhang, Q.-M., Ishikawa, N., Nakahara, T., and Yonei, S. (1998) Escherichia coli MutY protein has a guanine-DNA glycosylase that acts on 7, 8-dihydro-8-oxoguanine: guanine mispair to prevent spontaneous G: C→C: G transversions. Nucleic Acids Res. 26, 4669–4675.
- Zhang, Q.-M., Sugiyama, H., Miyabe, I., Matsuda, S., Kino, K., Saito, I., and Yonei, S. (1999) Replication in vitro and cleavage by restriction endonuclease of 5-formyluracil- and 5-hydroxymethyluracil-containing oligonucleotides. Int. J. Radiat. Biol. 75, 59–65.
- Zhang, Q.-M., Miyabe, I., Matsumoto, Y., Kino, K., Sugiyama, H., and Yonei, S. (2000) Identification of repair enzymes for 5-formyluracil in DNA: Nth, Nei and MutM proteins of Escherichia coli. J. Biol. Chem. 275, 35471–35477.
- Zhang, Q.-M., Hashiguchi, K., Kino, K., Sugiyama, H., and Yonei, S. (2003) Ntg1 and Ntg2 proteins as a 5-formyluracil-DNA glycosylases/AP lyases in Saccharomyces cerevisiae. Int. J. Radiat. Biol. 79, 341–349.
- Zharkov, D. O. (2008) Base excision DNA repair. Cell. Mol. Life Sci. 65, 1544–1565.