Review
Genetic abnormalities in Fibrodysplasia Ossificans Progressiva
2012 Volume 87 Issue 4 Pages 213-219
Details
2012 Volume 87 Issue 4 Pages 213-219
Fibrodysplasia ossificans progressiva (FOP), characterized by congenital malformation of bones, is an autosomal dominant disorder. This is a rare genetic disorder and its worldwide prevalence is approximately 1/2,000,000. There is no ethnic, racial, gender, or geographic predilection to FOP. It is regarded as one of the intractable disorders, which is not only an extremely disabling disease but also a condition of considerably shortened lifespan. Although the genetic defects of FOP are not completely known, several clinical and animal model studies have implicated that mutations in bone morphogenetic proteins, their receptors, and activin receptor type IA (ACVR1) genes are associated with FOP primarily. The noggin (NOG) gene has also been reported in some studies. In most of the cases of FOP, the mutation was found as ‘de novo’ however there is paternal age effect on mutations. Unfortunately, at present there is no efficient treatment for FOP. The recent discoveries of genetic basis of FOP provide a clue to the underlying pathophysiology and potential therapy. This review article focuses on the genetic mutations in FOP, their usage as diagnostic markers, and possible target specific drug development to treat FOP patients.
Fibrodysplasia ossificans progressiva (FOP) is a rare autosomal dominant disorder of dysregulated cellular differentiation, which is characterized by congenital malformation of the great toes during embryonic skeletal development and by progressive heterotopic endochondral ossification of the connective tissue post-natally that forms qualitatively normal bone in characteristic extra-skeletal sites (Ratbi et al., 2010; Pignolo et al., 2011; Chakkalakal et al., 2012). Extra-skeletal bone formation associated with inflammation preceding the osseous conversion usually begins in the first decade, predominantly in the head, neck, and shoulders (Kartal-Kaess et al., 2010). Although the general phenotype of FOP, including the presence of congenital malformations of the great toes, is constant among individuals, there is wide variation in the severity of the disorder (Shore et al., 1997). Children, who have FOP, appear normal at birth, except for congenital malformations of the great toes. During the first decade of life, sporadic episodes of painful soft tissue swellings (flare-ups) occur which are often precipitated by soft tissue injury, intramuscular injections, viral infection, muscular stretching, falls or fatigue. These flare-ups transform skeletal muscles, tendons, ligaments, fascia, and aponeuroses into heterotopic bone, rendering movement impossible for the children because of the calcification of the muscles and ankylosis of all the joints (Dzukou et al., 2005; Pignolo et al., 2011). Individuals with FOP are also at a risk for hearing loss and that the type of loss is predominantly conductive in nature (Levy et al., 1999).
The worldwide prevalence of FOP is approximately 1/2,000,000. There is no ethnic, racial, gender, or geographic predilection to FOP. It occurs sporadically and is transmitted as a dominant trait with variable expression and complete penetrance. The disease symptoms appear in early life, and its course is unavoidably progressive (Mahboubi et al., 2001).
In 2007, FOP was indicated as one of the intractable disorders by the Ministry of Health, Labor and Welfare of Japan (Katagiri et al., 2010). It is not only an extremely disabling disease but also a condition of considerably shortened lifespan (Kaplan et al., 2010). The median lifespan is approximately 40 years of age. Most patients become wheelchair-bound by the end of the second decade of life, and commonly die of complications of thoracic insufficiency syndrome (Pignolo et al., 2011). Kaplan et al. (2010) also suggested cardio-respiratory failure from thoracic insufficiency syndrome and pneumonia as the most common causes of death in patients with FOP. They have verified the causes of FOP patient’s death with extensive medical records. Sixty deaths (thirty male and thirty female patients) were reported in International Fibrodysplasia Ossificans Progressiva community during a thirty-three-year-period. The median age at the time of death was forty years. The median lifespan estimated from the 371 individuals in the community who were alive and the sixty who had died was fifty-six years (Kaplan et al., 2010).
FOP is a rare genetic disorder, in which, mutations in some genes related to bone morphogenesis have been reported. However the genetic abnormalities in FOP are not clearly known. Connor and Evans (1982) found a point prevalence of 0.61 per million in the United Kingdom and gave a direct estimate of the mutation rate of 1.8 per million gametes per generation.
Although the genetic defects of FOP are not completely known, mutations in bone morphogenetic proteins (BMPs) and their receptors have been reported to be associated with FOP. BMPs are the members of a class of ancient, highly conserved signaling molecules, which play the major roles in embryonic axis determination, organ development, tissue repair, and regeneration in animals. Thus the BMPs are potent developmental morphogens that act in a concentration-dependent manner to specify cell fates in developing and regenerating systems (Kaplan and Shore, 1998). Several lines of evidence have suggested that BMP4 is involved in the pathophysiology of FOP (Kan et al., 2004).
Feldman et al. (2007) suggested that abnormal expressions of BMP4 and BMP5 proteins are associated with an array of human axial skeletal abnormalities, of which characteristics are similar to FOP. Gannon et al. (1997) indicated that BMP2 and BMP4 defects are involved in the pathogenesis of FOP lesions. NF-kappaB is an inflammatory mediator, which plays an important role in developmental skeletogenesis and in suppression of BMP4 expression. Because of its multiple roles in inflammation, skeletogenesis, and BMP4 regulation, NF-kappaB is supposed to play the critical role in the pathogenesis of FOP (Ahn et al., 2003).
Nearly all cases of FOP are caused by the identical heterozygous single nucleotide substitution (c.617G>A; p.R206H) in the bone morphogenetic protein (BMP) type I receptor activin receptor type IA (ACVR1), also known as ALK2 (Chakkalakal et al., 2012; Shore, 2012). ACVR1 gene belonging to the transforming growth factor (TGF)-β superfamily (Song et al., 2012), consists of 139.417 kb, and was mapped to chromosome 2q23–24 (NCBI GenBank, 2012). Nakajima et al. (2007) reported the c.617G>A; p.R206H mutation in the ACVR1 gene in individuals with FOP of various ethnic groups, and they suggested that mutation in the ACVR1 gene is common and recurrent in the global population. Lucotte et al. (2009) analyzed the mutations in French FOP patients, and reported that 14 out of 27 (52%) individuals have c.617G>A mutation in the ACVR1 gene. Lee et al. (2009) performed the mutation analysis of the ACVR1 gene in 12 Korean patients diagnosed or suspected to have FOP. They found all patients had a de novo heterozygous point mutation of c.617G>A; p.R206H in ACVR1. Sun et al. (2009) identified the presence of a single heterozygous c.617G>A (p.R206H) mutation in the ACVR1 gene in two Chinese FOP patients. Zhou et al. (2008) reported a 3-year-old Chinese girl with typical FOP characteristics, in whom the mutation c.617G>A; p.R206H in ACVR1 was identified. In another study by Du et al. (2010), mutation at the same position (c.617G>A; p.R206H in ACVR1) was confirmed in a 4-year-old Chinese FOP patient. Lin et al. (2006) reported a 3-year-old Taiwanese girl with FOP characteristics and mutation in c.617G>A; p.R206H position. The mutation c.617G>A causing p.R206H amino acid substitution, lies within a highly conserved glycine-serine (GS) activation, domain close to the border with the protein kinase domain. Generally extracellular BMPs bind to BMP type I and type II receptors, leading to GS domain phosphorylation and hence activation of downstream pathways, but in the presence of the p.R206H mutation, ACVR1 (BMP type I receptor) become mildly constitutively active, that is they are able to activate downstream signaling in the absence of ligand stimulation (Gregson et al., 2011). Whyte et al. (2012) reported a missense defect in another position of ACVR1 gene (c.974G>C; p.G325A) that predicted a conservative (mild) amino acid change within the kinase domain of ALK2. Further two unique mutations (c.605G>T and c.983G>A) in ACVR1 in two FOP patients with some atypical digit abnormalities and other clinical features, have also been reported by Petrie et al. (2009). A group of 16 Brazilian individual with FOP phenotypes was screened by Carvalho et al. (2010), all having the classical mutation c.617G>A; p.R206H. In this study, one woman had mutation in the position of c.983G>A (p.G328E). Carvalho et al. (2010) suggested that variant FOP phenotypes are associated with specific mutations in ACVR1 gene. Ratbi et al. (2010) reported the case of a Moroccan patient with FOP carrying a rarely occurring mutation at c.774G>T of ACVR1 gene. Bocciardi et al. (2009) reported the c.617G>A mutation, leading to the p.R206H substitution in the ACVR1 gene in 15 out of 17 Italian patients. In two patients, they found a novel mutation c.774G>C; p.R258S substitution (Bocciardi et al., 2009). Furuya et al. (2008) reported a 62-year-old man with slowly progressive FOP, and bearing a novel mutation in ACVR1. He had a difficulty in moving his shoulder since age of 10 years due to contraction of the shoulder joint. The symptoms progressed slowly, and he could not walk at the age of 36 years and was bedridden at the age of 55 years. He also showed rigid spine, baldness, sensorineural hearing loss, and hypodactyly accompanied by abnormal ectopic ossification. Analysis of ACVR1 and its cDNA revealed that the patient had a heterozygous mutation at c.1067G>A (p.G356D), which was a de novo mutation (Furuya et al., 2008). The causative mutations in ACVR1 have been summarized in Table 1.
| Gene name | Mutations | References |
| ACVR1 | c.605G>T (p.R202I) | Petrie et al. (2009) |
| c.617G>A (p.R206H) | Lin et al. (2006); Bocciardi et al. (2009); Sun et al. (2009); Lee et al. (2009); Du et al. (2010); Chakkalakal et al. (2012) | |
| c.774G>T (p.R258S) | Ratbi et al. (2010) | |
| c.774G>C (p.R258S) | Bocciardi et al. (2009) | |
| c.974G>C (p.G325A) | Whyte et al. (2012) | |
| c.983G>A (p.G328E) | Petrie et al. (2009); Carvalho et al. (2010) | |
| c.1067G>A (p.G356D) | Furuya et al. (2008) | |
| NOG | c.271G>T (p.G91C) | Sémonin et al. (2001) |
| c.274G>C (p.G92R) | Sémonin et al. (2001); Lucotte et al. (2009) | |
| c.275G>A (p.G92E) | Sémonin et al. (2001); Lucotte et al. (2009) | |
| c.276G>A (p.G92G) | Lucotte et al. (2009) | |
| c.283G>A (p.A95T) | Fontaine et al. (2005); Lucotte et al. (2009) | |
| delta 42 (Removal of 14 amino acids from 87–100 position of protein) | Sémonin et al. (2001); Lucotte et al. (2009) |
Where, c=cDNA; A=adenine, G=guanine, T=thymine, C=cytosine.
ACVR1 is normally inactive until it binds to extracellular BMP (Song et al., 2012). BMPs interact with specific BMP receptors (BMPRs) that phosphorylate downstream effector molecules. Several combinations of the type I and type II BMPRs mediate the BMP-4 signaling pathway. For example, after ligand binding, constitutively activated type II BMPRs attract and phosphorylate type I BMPRs. Then the activated type I BMPRs (for example, ACVR1), subsequently phosphorylate the intracellular proteins of at least two interacting signaling cascades: the canonical Smad pathway and the p38 mitogen-activated protein kinase (p38 MAPK) pathway. Both pathways regulate nuclear transcriptional activity through the cofactor association in a cell and in developmental-specific context (Kaplan et al., 2006). ACVR1 has been found to be able to cause endothelial-to-mesenchymal transition (EndMT) in endothelial cells, which might cause the formation of FOP lesions. FOP-associated mutant ACVR1-p.R206H renders ACVR1 constitutively active and increases the phosphorylation of downstream Smad1 effector proteins, which stimulates its phosphorylation within the GS domain. This activates the ACVR1 to recruit and phosphorylate Smads 1, 5, and 8 within the cell. Subsequently, these form heteromeric complexes with the co-regulatory Smad4 and accumulate in the nucleus. Once phosphorylated, by directly binding to DNA and interacting with different transcriptional co-activators or co-repressors, Smads regulate target gene transcription (Song et al., 2012). The mutant ACVR1-p.R206H sensitizes mesenchymal cells to BMP-induced osteoblast differentiation, and stimulates new bone formation, i.e., the mutant p.R206H ACVR1 activates BMP signaling in the absence of BMP ligand and mediates BMP-independent chondrogenesis (Shen et al., 2009) (Fig. 1). Song et al. (2012) investigated whether miR-148a directly targets ACVR1 mRNA by reporter gene assays and mutational analysis at the miRNA binding sites, and inhibited ACVR1 both at the protein level and mRNA level. They suggested that miR-148a is an important mediator of ACVR1, thus offering a new potential target for the development of therapeutic agents against FOP (Song et al., 2012). According to Song et al. (2010), the p.R206H mutant showed a decreased binding affinity for FKBP1A/FKBP12, a known safeguard molecule against the leakage of transforming growth factor (TGF)-beta or BMP signaling. The decreased binding affinity of FKBP1A to the mutant p.R206H ACVR1 resulted in leaky activation of the BMP signal, and moreover, it decreased steady-state p.R206H ACVR1 protein levels.
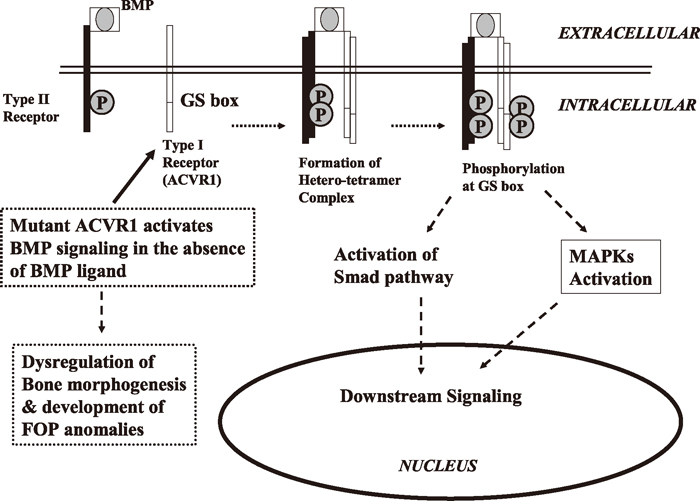
Molecular mechanism of BMP-ACVR1 signaling and Fibrodysplasia Ossificans Progressiva. BMPs bind to and activate the type II receptor, which attracts and phosphorylates type I receptor (ACVR1). The activated type I receptor, subsequently phosphorylates the intracellular proteins of at least two interacting signaling cascades: the canonical Smad pathway and the p38 mitogen-activated protein kinase (p38 MAPK) pathway. Both pathways regulate nuclear transcriptional activity for bone morphogenesis in developmental-specific context (Kaplan et al., 2006; Song et al., 2012). Mutations in ACVR1 activate BMP signaling in the absence of BMP ligand, and mediate BMP-independent chondrogenesis (Shen et al., 2009), which develop FOP phenotypes.
The noggin gene (NOG), located on chromosome 17q22, is another important mutation prone candidate gene for FOP (Lucotte et al., 2000). Fontaine et al. (2005) reported mutation at nucleotide position 283 (c.283G>A) in this gene in a French family burdened with FOP. In another study, mutations in the nucleotide positions of c.271G>T, c.274G>C, c.275G>A of NOG have been reported in three Spanish families burdened with FOP (Sémonin et al., 2001). All three mutations, as well as the Delta 42 deletion resulted in the alteration of the portion of the NOG gene at the positions 265–282, which encode for the N-myristoylation site at residues 89-GGGGGA-94 that is important for the functioning of gene product (Sémonin et al., 2001). Lucotte et al. (2009) reported five NOG mutations (delta 42, c.274G>C, c.275G>A, c.276G>A, and c.283G>A) in 7 individuals out of 27 French FOP patients. The causative mutations in NOG have been summarized in Table 1.
It is debatable whether there is any paternal effect on FOP. Zhou et al. (2008) reported a Chinese child with the mutation c.617G>A in ACVR1. They suggested it as a de novo mutation, as no such type of mutation was found in the child’s parents. Mutation in c.617G>A in ACVR1 gene in patient but not in parents of Chinese individual has been further confirmed by Du et al. (2010). De novo mutation in the ACVR1 gene was also suggested in a Taiwanese girl by Lin et al. (2006). But, in a French FOP family, c.283G>A mutation in the NOG gene was found to be transmitted in the family (in the heterozygote form) by the affected mother to her two affected children (Fontaine et al., 2005).
The paternal age may have influence on FOP. In 1979, Rogers and Chase surveyed on 42 patients with FOP to assess their clinical status. They found a significant paternal age effect. The confirmation of a paternal age effect in FOP would support the proposal that it usually occurs as a new mutation (Rogers and Chase, 1979). Some years later, in 1982, Connor and Evans investigated 44 FOP patients in the United Kingdom but found no evidence for genetic heterogeneity in this series. All patients represented fresh gene mutations and all patients were unable to inherit their disease allele to the next generation. According to their report, a significant paternal age (>30 years) effect was evident for these new mutations. They reported that if the potential father is over 50 years of age then the risk is probably seven-fold that of a father of 30 years (Connor and Evans, 1982).
The emergence of relevant animal models for FOP is crucial for understanding the disease. A sporadic FOP-like condition was described in domestic housecats, but no known living animal was found available for further study. The first animal model that provided a clue to the cause of FOP was recognized in the decapentaplegic (dpp) mutants of Drosophila, and predicted the role of BMP-signaling in the pathophysiology of FOP (Kaplan et al., 2005). Kaplan et al. (1990) discovered an array of developmental gradients (characteristic patterns of disease expression) in D. melanogaster similar to the developmental anomalies induced by pleiotropic mutations of the dpp locus. Among the developmental anomalies in model, expression of endochondral bone formation is the basis for limb formation in embryogenesis, longitudinal bone growth in postnatal life, and local bone regeneration (fracture healing) following injury. The protein encoded by the dpp locus is a member of the transforming growth factor-beta (TGF-beta) family, which shares 75% sequence homology with the c-terminal region of BMP-2A, BMP-2B (also members of the TGF-beta family). The sequence identity of BMP genes between man and Drosophila (although large evolutionary distance) suggests that these genes may have been derived from common ancestral gene, and they are evolutionarily conserved. The developmental similarities between dpp in Drosophila and FOP in human suggest dpp mutant flies as a useful model for the study of FOP (Kaplan et al., 1990). Kan et al. (2004) developed a unique transgenic mouse line that overexpresses BMP4 under the control of the neuron-specific enolase (NSE) promoter, which develops a FOP-like phenotype. Mating of these animals with transgenic animals that overexpress the BMP inhibitor NOG, prevents the disorder, and confirmed the role of BMP4 in the pathogenesis of the disease. This remarkable animal model provided a good opportunity to further study the role of the BMP signaling pathway in heterotopic ossification and to improve the understanding of the clinical aspects of FOP (Kan et al., 2004).
Most patients with FOP are being misdiagnosed early in life before the appearance of heterotopic ossification and undergo diagnostic procedures such as biopsy that can cause lifelong disability. Genetic studies could be performed as a potent diagnostic way. Once diagnosed, patients should be advised in order to avoid unnecessary traumas, surgical procedures, biopsies, intramuscular injections and vaccinations (Kaplan et al., 1993; Ulusoy, 2012). It was found that incorrect diagnoses were given initially to 87% of individuals with FOP. This high rate of incorrect diagnostic errors occurred worldwide, regardless of ethnicity, geographic background, or misdiagnosing physician’s specialty. In FOP, the astonishing rates of diagnostic errors and inappropriate invasive medical procedures likely result from lack of physician awareness because of failure of information transfer (Kitterman et al., 2005). Zaghloul et al. (2008) presented a child with the classic features of previously undiagnosed FOP who developed a paraspinal soft-tissue mass after a lumbar puncture for a fever workup. Excision of the mass resulted in a massive inflammatory response leading to progression of heterotopic ossification (Zaghloul et al., 2008). Pediatricians should be aware of the early diagnostic features of FOP, even before the appearance of heterotopic ossification. Detection of c.617G>A in ACVR1 gene is being suggested as potent diagnostic marker for early diagnosis of FOP, which may help in early treatment. And obviously, early treatment will help to avoid the factors of aggravation, slow the progression of the disease and provide the children with improved quality of life (Dzukou et al., 2005).
Unfortunately, at present there is no efficient treatment for FOP. Bisphosphonates and corticosteroids are only beneficial drugs during the flares (Dzukou et al., 2005). A brief 4-day course of high-dose of corticosteroids, started within the first 24 hours of a flare-up, may help to reduce the intense inflammation and tissue edema seen in the early stages of the disease (Pignolo et al., 2011). Chemical inhibitors to the pathogenic ALK2 receptors, for example, Dorsomorphin (6-[4-(2-Piperidin-1-yl-ethoxy)phenyl]- 3-pyridin-4-yl- pyrazolo [1,5-a]pyrimidine), are considered as possible medical agents for FOP, but their adverse effects on normal ALK2 and other receptors cannot be excluded (Takahashi et al., 2012). However, a treatment strategy for FOP using allele-specific RNA interference (ASP-RNAi) has been designed recently, which showed modified small interfering RNAs (siRNAs) conferring allele-specific silencing against disease-causing ALK2 mutants found in FOP, without affecting normal ALK2 allele (Takahashi et al., 2012; Kaplan et al., 2012).
The recent discoveries of genetic basis of FOP, like the detection of overproduction of BMP-4 in lesional cells and lymphocytic cells of affected patients provide a clue to the underlying pathophysiology and potential therapy (Mahboubi et al., 2001). The discovery of the FOP related genes established a critical milestone in understanding FOP, which reveals a highly conserved target for drug development in TGF-beta/BMP signaling pathway, and compels therapeutic approaches for the development of small molecule signal transduction inhibitors of ACVR1/ALK2. Effective therapies for FOP, and possibly for a vast array of more common conditions of heterotopic ossification, may potentially be based on future interventions that block ACVR1/ALK2 signaling (Kaplan et al., 2007, 2008).
Although FOP is a rare genetic disorder, it is painful and life threatening. Recently, more emphasize has given for the research on this disease. Some genetic mutations have been confirmed, which may be used as molecular or genetic markers for the early diagnosis of the disease. Identification of these genetic abnormalities has also shown some promises for the development of target specific drugs and future treatment.
This study was funded by the project of Changsha City Bureau of artificial bone research for the development of drug with new composite materials (grant no- K1101025-31). We are thankful to Dr. Md. Asaduzzaman Khan, Department of Biochemistry, Central South University for his critical suggestions during manuscript preparation and editing.