ABSTRACT
We analyzed PCR-amplified carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (CAD) gene fragments from 146 Bombyx mori native strains and found extremely low levels of DNA polymorphism. Two haplotypes were identified, one of which was predominant. CAD haplotype analysis of 42 samples of Japanese B. mandarina revealed four haplotypes. No common haplotype was shared between the two species and at least five base substitutions were detected. This result was suggestive of low levels of gene flow between the two species. The nucleotide diversity (π) scores of the two samples differed markedly: lower π values were estimated for B. mori native strains than Japanese B. mandarina. We further analyzed 12 Chinese B. mandarina derived from seven areas of China, including Taiwan. The results clearly indicated that the π score was ~80-fold greater in Chinese B. mandarina than in B. mori. The extremely low level of DNA polymorphism in B. mori compared to its wild relatives suggested that the CAD gene itself or its tightly linked regions are possible targets for silkworm domestication.
INTRODUCTION
The mitochondrial cytochrome C oxidase I (COI) gene has been used extensively to study various aspects of phylogenetics and population genetics of insects (e.g., Aoki et al., 2008; de Jong et al., 2011). COI nucleotide sequence variation analysis has been used to classify the domestic silkworm, Bombyx mori, and to illustrate the genetic relationship of B. mori and its wild relative B. mandarina in Japan (Yukuhiro et al., 2011). Although the two species have a different number of chromosomes (B. mori has 28 whereas Japanese B. mandarina has 27; Kawaguchi, 1928; Nakamura et al., 1999), they generate fertile hybrids. Yukuhiro et al. (2011) demonstrated a significant genetic divergence between the COI nucleotide sequences of the two species, a result that is concordant with studies by Yukuhiro et al. (2002) and Pan et al. (2008). Moreover, this finding suggests that little gene flow occurs between B. mori and B. mandarina in Japan (Yukuhiro et al., 2011).
Despite the extensive and successful use of mitochondrial COI nucleotide sequences, it is important to analyze nuclear genes to explore phylogenetics and population genetics of organisms because most events of adaptive evolution are associated with molecular changes in nuclear genes. In insect species, several genes, including “carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase” (CAD) and DOPA decarboxylase (ddc), are frequently used for phylogenetic analysis and species identification (e.g., Regier et al., 2008). As described above, mitochondrial COI-based analyses have been conducted in B. mori and Japanese B. mandarina (Yukuhiro et al., 2011). However, the genetic relationship between B. mori strains and between B. mori and B. mandarina requires further elucidation using nuclear genes to assess how B. mori has affected the genetic background of Japanese B. mandarina through gene flow.
In the present study, we analyzed CAD gene fragment sequences from B. mori and B. mandarina inhabiting China and Japan to assess their genetic variation. The CAD gene of B. mori is located on the tenth chromosome. In this study, we designed a novel combination of PCR primers to stably obtain amplification products.
Amplified CAD fragments of the B. mori native strains housed in the National Institute of Agrobiological Sciences, Japan, showed extremely low genetic variation, wherein only one segregating site was found. In contrast, three variable sites (including one highly polymorphic site) were detected in 42 B. mandarina in Japan. The level of nucleotide diversity was about 12-fold greater than that of the B. mori native strain set. Furthermore, we identified at least five nucleotide substitutions between the two CAD haplotype sequences of B. mori native strains and four haplotype sequences in B. mandarina. This finding is consistent with that of Yukuhiro et al. (2011), which indicated that little gene flow occurred from B. mori to Japanese B. mandarina.
We investigated the genetic variation in Chinese B. mandarina, which is a probable ancestor of B. mori. Based on comparative analysis of B. mori and Chinese and Japanese B. mandarina, we confirmed a significant decrease in genetic variation in the CAD sequence of B. mori. Recently, Yu et al. (2011) and Guo et al. (2011) reported possible gene targets for silkworm domestication. In these reports, the authors employed the significantly decreased level of genetic variation in B. mori genes or regions compared to those in Chinese B. mandarina as a criterion for the domestication-targeted gene. For example, Guo et al. (2011) noted that the Defensin A (Def A) gene of B. mori, which encodes an antibacterial peptide involved in innate immunity (Tanaka et al., 2008), underwent artificial selection during the domestication process based on the fact that the nucleotide diversity score decreased to about 3% of that of Chinese B. mandarina, combined with coalescent simulation work. Guo et al. (2011) also indicated that the rates of reduction in domesticated silkworm diversity relative to wild B. mandarina ranged from 33% to 49%.
These reports suggest that strong artificial selection occurred during the domestication process because genetic polymorphisms at several loci have been maintained in B. mori (e.g., Kim et al., 2008; Yu et al., 2011). Xia et al. (2009) identified possible target genes of silkworm domestication by analyzing 29 B. mori and 11 B. mandarina individuals. However, the CAD gene was not present in their list of candidate genes. In this report, we discuss the different population structures of B. mori, Chinese B. mandarina, and Japanese B. mandarina based on the mitochondrial COI gene and nuclear CAD gene.
MATERIALS AND METHODS
Genomic DNA samples of native silkworm strains
B. mori DNA samples used in this study were the same as those described in Yukuhiro et al. (2011), except for one genomic DNA sample. These DNA samples were originally used in Tomita et al. (1997). We analyzed 48 Japanese, 49 Chinese, 28 European, and 21 molting strains. The European samples contained 26 native and two improved strains, whereas the other three strain classes contained exclusively native strains. A list of strains used in this study is shown in Table 1. The B. mori DNA samples were derived from multiple individuals per strain to maintain as much genetic variation as possible.
Table 1. List of silkworm strains used in this study
| Japanese strains | Chinese strains | European strains | Molting strains |
|---|
| Akajuku | Amoi moricaud | Yellow cocoon of Asia | Chojo |
| Akako | Benishina | Ascoli | Choyo |
| Aojuku (A) | Segregated white | Yellow cocoon of Ascoli | European No. 7 trimolter |
| Aojuku (Ichinomiya) | Chahan | Bagdad | Trimolter of Kan |
| Agunisan | C spherical cocoon | 91 Bagdad | Trimolter of Korai |
| Chikuma | Daiento | Pione | Menyo |
| Chusuotsu | Hekiren | Blanc des C’evennes | Sainan |
| Dainyorai | Hiko | Ginto | Sanko |
| Datenishiki | Kaijo | Gubbio | Trimolting white |
| Himeko | Kanko | Golden pink | Trimolter of Santo |
| Hinode (Odaka) | Kankoshaken | Black moth | Sen No. 3 |
| Kakumata (Aramomi) | Kansen | Maiella·Zebra | Golden yellow cocoon of Shisen |
| Kokin | Golden yellow | Yellow cocoon of Maruke | Trimoter of Shisen |
| Kinkoshu | Knobbed | No. 500 | Kyokuko |
| Kinshoku (Maebashi) | Kohoku | No. 65 | OK39 |
| Koishimaru | Kuroko (Shimane) | Rosa | VC1 |
| Koishimaru (Watari) | Kyugyoku | S No. 9 | Chogosan No. 1 |
| Kuniichi | Nanko | Spain | Pentamolting white |
| Matamukashi | Oha | St. Julien | Pentamolting black |
| Murasakiko | Seikyo | Szegsard | Pentamolter of Kyushu Univ. |
| Onichijimi | Sekko | Yellow cocoon of Turkey | N·S |
| Ryukyu multi-pupal cocoon (Kiaya) | Shakugen | Var | |
| Ryukyu multi-pupal cocoon (Shizuoka) | Chinese white cocoon | Y4 | |
| Sagami | Shinshocho | y35 | |
| Sakurahime | Shokei | y39 | |
| Seihaku (Shizuoka) | Shuko | Yuhaku | |
| Sekaiichi | S No. 1 | European No. 16 (Old) | |
| Tadamiko | Tenmon | European No. 16 (New) | |
| Tenryuseihaku | Tokuishin | | |
| Wako | Tsunoshina | | |
| Aojuku (Hanyuda) | Uryu | | |
| Chiyozuru | Y10 | | |
| Chusu | Yoko | | |
| Kinshoku (Matsumoto) | Daiankyo | | |
| Kojiki | Daikojo | | |
| Kunitomi | Daizo (Nagano) | | |
| Kuroko (Takahashi) | Daizo (Matsumura) | | |
| Kuroko | Hakuyoen | | |
| Nihonichi | Kinkomaru | | |
| Daiaojuku | Kosetsu | | |
| Onishiki | Striped | | |
| Seihaku (Yamagata) | Shoka | | |
| Shohaku (Maebashi) | Shoko | | |
| Shohaku (Taketoyo) | 209AB | | |
| Tamanashi kasuri | Low 209 | | |
| Tanegashima | 210 | | |
| Tsunomata (Shizuoka) | 213 | | |
| 214 | | |
| 216 | | |
Genomic DNA was extracted from the Japan-inhabiting wild mulberry silkmoth, B. mandarina from 17 locations in Japan (Table 2) using two different procedures: genomic DNA isolation from the whole adult moth using IsoTissue (Nippon Gene, Tokyo, Japan), according to the manufacturer’s instructions, or DNA isolation from a small portion of the adult moth body using DNAzol (Life technologies, Carlsbad, California, USA), again according to the manufacturer’s instructions, followed by purification with PI-1100 (Kurabo, Osaka, Japan).
Table 2. Numbers and sampling locations of Japanese
B. mandarina used in this study
| Locations (Prefectures) | Number of samples |
|---|
| Hakodate (Hokaido) | 4 |
| Morioka (Iwate) | 4 |
| Nanyo (Yamagata) | 1 |
| Fukushima (Fukushima) | 1 |
| Tamura (Fukushima) | 3 |
| Hirono (Fukushima) | 1 |
| Mt.Tsukuba (Ibaraki) | 2 |
| Ami (Ibaraki) | 1 |
| Shimonita Shimokosaka (Gunma) | 3 |
| Shimonita Minaminomaki (Gunma) | 1 |
| Shimonita Shiraidaira (Gunma) | 4 |
| Shimonita Uchiyamatoge (Gunma) | 4 |
| Saku Uchiyamatoge (Nagano) | 3 |
| Saku Ohtsuki, Kuroda (Nagano) | 1 |
| Kobuchizawa (Yamanashi) | 4 |
| Shizuoka (Shizuoka) | 3 |
| Shimaisobe (Mie) | 2 |
| Total | 42 |
Nucleotide sequences of the two CAD-specific primers, CAD2055F and CAD2702R, were:
CAD2055F GRGTAACYACAGCCTGTTTTGAAC
CAD2702R CCTATTCTATACACADCTGAACC
CAD2055F was based on CAD743nF described by Regier et al. (2008) with modifications to adapt to the B. mandarina CAD cDNA sequence with Genbank accession number EU032655. We originally prepared CAD2702R to correspond to the B. mandarina CAD cDNA sequence with Genbank accession number EU032655.
Reaction mixtures contained 1 × Ex Taq buffer (TaKaRa, Kyoto, Japan), 1.0 mM dNTP, 0.2 mM each primer, 0.5 unit of Ex Taq (TaKaRa), and approximately 5 ng of genomic DNA in a 20 μl reaction mixture. Cycling conditions were as follows: 2 min incubation at 96℃ and 40 cycles of 15 s at 96℃, 15 s at 44℃, and 1 min at 68℃, followed by a 2 min incubation at 68℃. We confirmed amplification by agarose gel electrophoresis followed by detection using Gel Red (Biotium, Hayward, CA, USA) staining. This amplicon was 646 bp long excluding sections corresponding to the two primers and part of the exonic sequence.
DNA sequencing and separation of the two sequences in diploid DNA data
Prior to DNA sequencing, we excluded the remaining primers and dNTPs by treatment with ExoSAP-IT (USB Corp., Cleveland, Ohio, USA), according to the manufacturer’s protocol. We used the ABI BigDye Terminator ver. 3.1 (Applied Biosystems, Foster City, CA, USA) for DNA sequencing. The sequencing reaction mixture (10 μl) contained the following components: 1.5 μl 5× buffer (Applied Biosystems), 1 μl Premix (Applied Biosystems), 0.1 μl of 20 μM primer for sequencing, 5.4 μl of dH2O, and 2.0 μl of purified PCR product. Cycling conditions were as follows: 2 min incubation at 96℃ followed by 35 cycles at 96℃ for 15 s, 50℃ for 10 s, and 60℃ for 4 min. Upon completion of the sequencing reaction, we purified the sequencing products on a Sephadex column. We used the Applied Biosystem 3730 DNA analyzer (Applied Biosystems) to detect sequencing signals. We used the CAD2702R primer for the direct sequencing. Most one-directional sequencing reactions yielded stable sequencing signals with little noise. However, the initial points of the sequences to be read were variable. Therefore, we analyzed the 567 bp region of the 646 bp amplicon and the 79 bp 3’ part of amplicon was excluded from analysis.
The nucleotide sequences were edited using ATGC (Genetyx Co., Tokyo, Japan) followed by DNA sequence alignment using Clustal W (Thompson et al., 1994) in MEGA 5 (Tamura et al., 2011). Because we identified a few overlapping peaks in the DNA sequences in the B. mori and Japanese B. mandarina data set, we separated the two sequences of the diploid DNA data using the Haplotype Reconstruction option in DnaSP version 5.0 (Librado and Rozas, 2009).
When conducting DNA sequencing of the CAD fragments of 12 Chinese B. mandarina samples (Table 3), we first directly sequenced the PCR product. As we frequently detected multiple overlapping peaks in DNA sequence data of the single product, the product was defined as a heterozygote. To confirm two constitutive nucleotide sequences in a heterozygote, we subcloned the fragments into the pGEM-T vector (Promega, Fitchburg, Wisconsin, USA) using TA-cloning, according to manufacturer’s protocol. Following isolation of plasmid DNA by standard procedures, we performed DNA sequencing in the manner described above, except that the T7 primer corresponding to the pGEM-T vector was used instead of the CAD2702R primer. By comparing this sequence data with those obtained from the directed sequencing, we obtained a pair of CAD nucleotide sequences.
Table 3. Chinese
B. mandarina samples
| Samples | Location where samples were collected | Sex | Persons collecting samples | Haplotype combination |
|---|
| ZS1 | Ziyang, Sichuan | Female | Q. Xia | CAD_C_Bma1&1 |
| ZS2 | Ziyang, Sichuan | Male | Q. Xia | CAD_C_Bma1&1 |
| JJ1 | Jiangsu, Jiangsu | Female | Q. Xia | CAD_C_Bma1&2 |
| JJ2 | Jiangsu, Jiangsu | Male | Q. Xia | CAD_C_Bma1&3 |
| YY1 | Yunnan, Yunnan | Female | Q. Xia | CAD_C_Bma7&8 |
| YY2 | Yunnan, Yunnan | Male | Q. Xia | CAD_C_Bma2&9 |
| SS1 | Shandong, Shandong | Female | Q. Xia | CAD_C_Bma1&1 |
| XSi1 | Xichang, Sichuan | Female | Q. Xia | CAD_C_Bma10&12 |
| XSi2 | Xichang, Sichuan | Male | Q. Xia | CAD_C_Bma10&10 |
| XSh1 | Xi’an, Shaanxi | Nd* | M. Wang | CAD_C_Bma1&11 |
| MT1 | Miaoli, Taiwan | Nd | S. Gu | CAD_C_Bma4&5 |
| MT2 | Miaoli, Taiwan | Nd | S. Gu | CAD_C_Bma6&7 |
*Nd means that the sex of the sample individual was not determined.
Subsequently, DnaSp ver. 5.0 (Librado and Rozas, 2009) was used to detect segregating sites and identify constitutive haplotypes to estimate the population genetic parameters, including the number of segregating site (S), haplotype diversity (Hd) and its standard error, nucleotide diversity (π) and standard error, θw and its standard error, and the average number of nucleotide differences (k). The software was also used to conduct MK tests (McDonald and Kreitman, 1991) and Tajima D tests (Tajima, 1989). TCS 1.21 (Clement et al., 2000) was used to construct the maximum-parsimony haplotype network. Nucleotide substitution rates of the CAD and COI gene between Japanese B. mandarina and B. mori were estimated using MEGA 5 (Tamura et al., 2011). The COI nucleotide sequences used in this analysis were identical to those in Yukuhiro et al. (2011).
DNA sequences of B. mori CAD haplotypes reported here (CAD_Bmo1 and 2) are available in the DDBJ/EMBL/GenBank databases under accession numbers AB706309 and AB706315. The accession numbers of five haplotype sequences of Japanese B. mandarina CAD fragments are as follows: AB706310 for CAD_J_Bma1, AB706311 for CAD_J_Bma2, AB706312 for CAD_J_Bma3, AB731186 for CAD_J_Bma4. DDBJ/EMBL/GenBank accession numbers AB706316 to AB706327 were assigned to 12 Chinese B. mandarina CAD haplotype sequences, CAD_C_Bma1 to Bma12.
RESULTS
Segregating site and nucleotide diversity in B. mori native strains
We successfully isolated CAD fragments from 146 B. mori native strains by PCR. Following DNA sequencing and subsequent editing, we aligned 146 sequence pairs and separated each diploid sequence into two haploid sequences. In total, we prepared 292 B. mori CAD nucleotide sequences.
As mentioned above, the B. mori DNA samples originated from multiple individuals per strain. Therefore, if no overlapping peak was identified in the DNA sequence file, the strain was considered to be a homozygote. If an overlap was detected, the strain was defined as a heterozygote. Hereafter, we analyzed the sequencing results based on this criterion.
Upon analyzing 567 sites from 292 sequences, we detected only one segregating site at site 291 (Fig. 1A). The two resulting haplotypes were defined as CAD_Bmo1 and Bmo2; CAD_Bmo1 carried G at site 291, whereas CAD_Bmo2 carried A at this site (Fig. 1A). This nucleotide difference between the two haplotypes is synonymous and did not induce an amino acid change.
Of the 292 B. mori CAD nucleotide sequences, 284 were CAD_Bmo1 sequences and eight were CAD_Bmo2. There were 140 CAD_Bmo1 homozygotes, 4 CAD_Bmo1 and 2 heterozygotes, and 2 CAD_Bmo2 homozygotes. The Hd score was 0.0530 ± 0.0018 (Table 4).
Table 4. Population genetic parameters of
CAD fragments of
B. mori and
B. mandarina inhabiting Japan and China
| B. mori | B. mandarina inhabiting Japan | B. mandarina inhabiting China |
|---|
| No. strains or samples | 146 | 42 | 12 |
| No. sequences | 292 | 84 | 24 |
| S (No. segregating sites) | 1 | 3 | 14 |
| No. haplotypes | 2 | 4 | 12 |
| Hd (Haplotype diversity) | 0.053 | 0.567 | 0.844 |
| Standard error of Hd | 0.018 | 0.027 | 0.064 |
| π (Nucleotide diversity) | 0.00009 | 0.00112 | 0.00725 |
| Standard error of π | 0.00003 | 0.00009 | 0.00088 |
| πs (π for synonymous sites) | 0.00046 | 0.00447 | 0.03530 |
| πns (π for nonsynonymous sites) | 0.00000 | 0.00025 | 0.00000 |
| k (Average number of nucleotide differences) | 0.005 | 0.633 | 4.109 |
The π score at whole sites was estimated to be 0.00009 ± 0.00003, whereas this score was 0.00046 at synonymous sites (Table 4). θw was 0.00028 ± 0.00028 and Tajima’s D value was estimated to be –0.63513, which was not statistically significant (P > 0.10).
Segregating sites and nucleotide diversity in CAD fragments from Japanese B. mandarina
Using the same method performed on B. mori native strains, we examined CAD sequences of Japanese B. mandarina. We analyzed PCR products from 42 individuals (Table 2) and identified 84 CAD sequences with three segregating sites (Fig. 1, A and B). We detected four haplotypes, CAD_J_Bma1 to 4, of which the frequencies of the four haplotypes were highly variable. As shown in Table 5, CAD_J_Bma1 and 2 were present at almost equal frequencies and differed at site 189, resulting in a synonymous change (CAD_J_Bma1: C vs. CAD_J_Bma2: T). The haplotype CAD_J_Bma3 differed from CAD_J_Bma1 at site 187 (CAD_J_Bma1: A vs. CAD_J_Bma3: C). This nucleotide difference between CAD_J_Bma1 and 3 induced a conserved amino acid change from Ile to Leu. The haplotype CAD_J_Bma4 was a singleton and differed from CAD_J_Bma2 at site 162 where CAD_J_Bma4 had A and CAD_J_Bma2 had a G. This nucleotide change was synonymous. The CAD Hd score of Japanese B. mandarina was estimated to be 0.5670 ± 0.0270.
Table 5. Numbers and frequencies of
CAD haplotypes of 42 samples derived from Japanese
B. mandarina| Haplotypes | No. of fragments | Frequencies |
|---|
| CAD_J_Bma1 | 43 | 0.5119 |
| CAD_J_Bma2 | 35 | 0.4167 |
| CAD_J_Bma3 | 5 | 0.0595 |
| CAD_J_Bma4 | 1 | 0.0119 |
| Total | 84 | 1.0000 |
The π score was estimated to be 0.00112 ± 0.00009 at whole sites, while the score of synonymous sites was 0.00447, 10 times higher than that of B. mori. θw was 0.00106 ± 0.00065 and Tajima’s D value was estimated to be 0.10219, which was not statistically significant (P > 0.1; Tajima, 1989).
This π score was about 12-fold greater than that of B. mori (π = 0.00009 ± 0.00003). These results suggested that B. mori CAD sequences were under artificial selection during the silkworm domestication process.
Nucleotide substitution between B. mori and Japanese B. mandarina
Aligned sequence data sets of B. mori and Japanese B. mandarina showed five fixed base substitutions, all of which were synonymous (Fig. 1B). A maximum parsimony network clearly showed significant differentiation between the two types of sequence sets (Fig. 2). These results were consistent with mitochondrial DNA-based analyses (ND5: Yukuhiro et al. (2002); COI: Pan et al. (2008); Yukuhiro et al. (2011)) despite the lower variation in CAD. As described in Yukuhiro et al. (2011), the present findings suggest that little gene flow occurs between B. mori and Japanese B. mandarina, although fertile hybrids between them have been generated under artificial conditions.
Comparison of genetic variation levels in CAD fragments
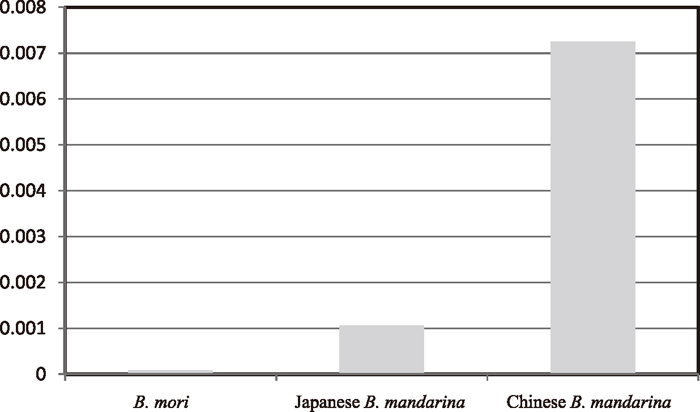
As described above, we detected a low level of π score in the CAD fragment among B. mori native strains. Compared with the B. mandarina samples, the π score was about 12 times lower than that of B. mandarina of Japan (Fig. 3). To examine the selection on CAD genes, we conducted a McDonald-Kreitman test (McDonald and Kreitman, 1991) using B. mori and Japanese B. mandarina CAD sequences, but no significant results were obtained.
Genetic variation in B. mori CAD compared to Chinese B. mandarina
We amplified and subsequently sequenced CAD fragments from 12 Chinese B. mandarina samples, as shown in Table 3. Following DNA sequence variation analysis, we identified 12 haplotypes (CAD_C_Bma1 to 12) based on 14 segregating sites (Fig. 1C). The number of haplotypes and their distribution among the samples are shown in Table 6.
Table 6.
CAD haplotype distribution of 24 Chinese
B. mandarina sequences
| No. sequences | Area |
|---|
| Jiangsu | Shandong | Miaoli | Xi’an | Xichang | Yunnan | Ziyang |
|---|
| CAD_C_Bma1 | 9 | 2 | 2 | | 1 | | | 4 |
| CAD_C_Bma2 | 2 | 1 | | | | | 1 | |
| CAD_C_Bma3 | 1 | 1 | | | | | | |
| CAD_C_Bma4 | 1 | | | 1 | | | | |
| CAD_C_Bma5 | 1 | | | 1 | | | | |
| CAD_C_Bma6 | 1 | | | 1 | | | | |
| CAD_C_Bma7 | 1 | | | 1 | | | | |
| CAD_C_Bma8 | 1 | | | | | | 1 | |
| CAD_C_Bma9 | 1 | | | | | | 1 | |
| CAD_C_Bma10 | 4 | | | | | 3 | 1 | |
| CAD_C_Bma11 | 1 | | | | 1 | | | |
| CAD_C_Bma12 | 1 | | | | | 1 | | |
| Total | 24 | 4 | 2 | 4 | 2 | 4 | 4 | 4 |
The estimated π score was 0.00725 ± 0.00088 (Table 4), which was about 80 times greater than that of B. mori (Table 4 and Fig. 3). The θw score was 0.00661 ± 0.00270. According to Guo et al. (2011), this result suggests that the CAD gene of B. mori may have experienced artificial selection during the silkworm domestication process. Although we conducted an MK-test, the G test could not be performed because of the lack of non-synonymous changes between B. mori and Chinese B. mandarina.
The B. mori CAD gene corresponds to the predicted gene BGIBMGA006816-TA in the Silkworm Genome Database. Although Xia et al. (2009) generated a list that included candidates for domestication genes in genomic regions of selective signals (GROSS), this gene was not included. However, the gene next to CAD, which was assigned as BGIBMGA006870-TA, was found in the list (SWGROSS0143). The two genes were approximately 7 kb apart. Xia et al. (2009) and Guo et al. (2011) observed rapid decay in linkage disequilibrium (LD); for example, Guo et al. (2011) showed that LD rapidly decays within 200 bp. These results indicate that the reduced variation at the CAD gene may not be caused by BGIBMGA006870-TA-mediated sweep.
Relationship between B. mori, Chinese B. mandarina and Japanese B. mandarina
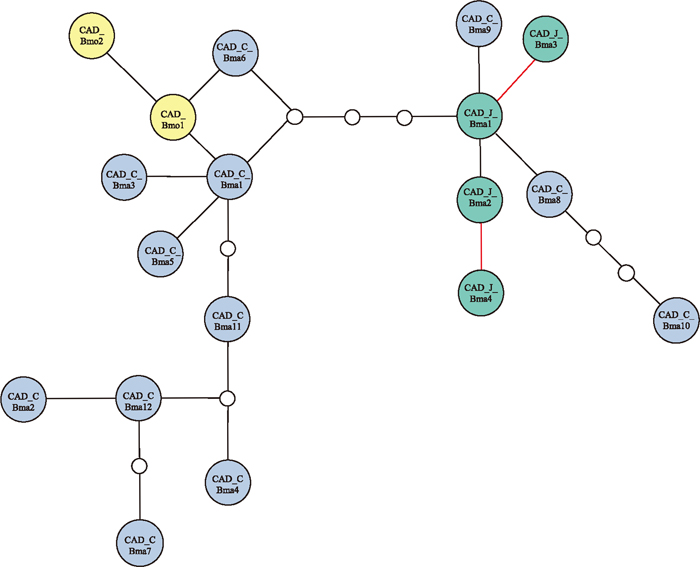
Since CAD haplotypes were identified in Chinese B. mandarina, we reconstructed a maximum parsimony network of 18 CAD haplotypes detected in this study (Fig. 2). The 18 haplotypes were classified into two clades: one included two haplotypes derived from B. mori and nine of Chinese B. mandarina, and the other included five Japanese B. mandarina haplotypes and three Chinese B. mandarina haplotypes. Considering the relationship between B. mori and Japanese B. mandarina, this network is consistent with that in Fig. 2. However, the fact that Chinese haplotypes were separated into two categories is suggestive of a high degree of genetic variability in Chinese B. mandarina, which may include Japanese B. mandarina.
DISCUSSION
In this study, we examined the DNA variation in CAD fragments of 146 B. mori native strains, 42 individuals of Japanese B. mandarina and 12 samples of Chinese B. mandarina derived from various areas of China, including Taiwan (Tables 1, 2, and 3, respectively).
At least five nucleotide substitutions (all of which were synonymous) were identified between B. mori and Japanese B. mandarina (Fig. 1, A and B; Fig. 2). This finding is consistent with results of the mitochondrial COI gene (Yukuhiro et al., 2011); that is, the two species shared no common haplotypes, suggesting that little gene flow occurs between them at the nuclear gene level. Whereas five nucleotide substitutions were found in CAD, 12 substitutions were confirmed in the mitochondrial COI coding sequence, which were also synonymous (Yukuhiro et al., 2011). Nucleotide substitution rates of CAD and COI between the two species were 0.0089 and 0.0170, respectively, based on results from the Jukes-Cantor method. The decreased rate of CAD nucleotide substitutions compared to that of COI is probably due to the lower mutation rate on the CAD gene relative to COI. That is, the mitochondrial COI synonymous mutation rate appears to be about two times higher than the CAD gene.
Recently, Osada and Akashi (2012) showed that the synonymous nucleotide changes in mitochondrial genes between Drosophila simulans and D. sechellia were only 1.2 to 2.5 times higher than in nuclear genes. The present estimate agrees with the results of Osada and Akashi (2012), suggesting that the mutation rates at mitochondrial genes in insects are not much different from those at nuclear loci, although this is based on only one species group.
Only two sites, 187 and 189, were found to be polymorphic in Japanese B. mandarina (Fig. 1), where more than two sequences of less frequent nucleotides were identified. The π scores at sites 187 and 189 were estimated to be 0.1131 and 0.4957, respectively. The nucleotide change at site 187 induced amino acid polymorphisms in a conserved manner; namely, CAD_J_Bma3 encoded Leu whereas CAD_J_Bma1, 2, and 4 encoded Ile. These results suggest that the CAD fragment used in this study is under strong functional constraint in the natural Japanese population. In addition, all nucleotide changes found in Chinese B. mandarina were synonymous, although the magnitude of variation in CAD nucleotide sequence was much higher than that of Japanese B. mandarina (Table 4). This result also supports the existence of a strict functional constraint on the CAD gene.
When compared with the mitochondrial DNA-based phylogenetic tree (Li et al., 2010) and the COI-based maximum parsimony network (Yukuhiro et al., 2011), the network of 18 CAD haplotype sequences in Fig. 2 formed a different network shape. Both mitochondrial gene-based relationships clearly showed that the clade (including B. mori) was separate from that of Japanese B. mandarina. The tree presented by Li et al. (2010) also demonstrated that the Chinese B. mandarina was located in a clade different from that of B. mori. In contrast, our deduced network showed that the two B. mori haplotypes belonged to a clade including most of the Chinese B. mandarina CAD haplotypes. These discrepancies might be derived from a difference in the effective population size (Ne) between nuclear and mitochondrial genes. That is, the Ne of the nuclear gene is four times higher than that of the mitochondrial gene. Therefore, a more pronounced geographic differentiation can be seen in the mitochondrial gene.
As shown in Fig. 3, a dramatic decrease in the π score was confirmed in the CAD gene of B. mori. Guo et al. (2011) demonstrated that a marked decrease in π value of B. mori Def A gene relative to that of Chinese B. mandarina supported that the Def A gene was under artificial selection during domestication. A similar result was presented by Yu et al. (2011). The present result is consistent with those of Guo et al. (2011) and Yu et al. (2011), suggesting that CAD is a candidate gene for silkworm domestication. As described above, the BGIBMGA006816-TA in the Silkworm Genome Database including the CAD gene was not found in the list of genes that represent good candidates for domestication genes (Xia et al., 2009) and therefore was considered to be a neutral gene for artificial selection (Xia et al., 2009). Although these results may provide a novel target gene for silkworm domestication, it should be noted that a gene found in the list (BGIBMGA006870-TA) is located about 7 kb from CAD. Guo et al. (2011) indicated that curves of LD decay rapidly within 200 bp. Therefore, the possibility of selective sweep on the CAD gene caused by the BGIBMGA006870-TA gene remains possible. Nevertheless, extensive surveys are required to examine nucleotide variation between the two genes as well as changes in the expression profiles of CAD between B. mori and B. mandarina to clarify whether the CAD gene is a target of silkworm domestication. Unfortunately, little information is available on the genomic structure and recombination rate in this region.
The π of CAD of Japanese B. mandarina was much higher than that of B. mori, but lower than that of Chinese strains. Although the sample size is not large, Chinese samples are regarded as representatives of the whole Chinese B. mandarina population because most of the range was covered by our sampling locations. In contrast, the π of B. mori pheromone biosynthesis activating neuropeptide (PBAN) (Guo et al., 2011) was 0.02282 ± 0.00408, higher than that of Japanese B. mandarina (0.00163 ± 0.00072), but similar to Chinese B. mandarina (0.0298 ± 0.0020). Comparing the period genes of Japanese and Chinese B. mandarina (Guo et al., 2011), the π of Japanese B. mandarina was lower than the Chinese strain.
These results suggest that, relative to the Chinese population, the effective population size of the Japanese population seems to be small. The π score obtained from an analysis of COI based on the large sample size of Japanese B. mandarina was also low, indicative of the small effective population size of Japanese B. mandarina and a short demographic history (Yukuhiro et al., in preparation). Incidentally, the higher π score of B. mori found in the PBAN gene suggests that the B. mori gene was under neutral conditions of artificial selection and maintained as much genetic variation as the original population.
In addition to analysis of Chinese B. mandarina originating from an extensive area within China, sampling and analysis of more Japanese B. mandarina populations will yield a better understanding of the genetic structure and relationship between Chinese and Japanese B. mandarina. Note that Fig. 2 suggested that CAD haplotypes found in the Shichong and Yunnan samples were clustered into the same clade as those of Japanese B. mandarina. Because a limited number of nucleotide sites (567 sites) have been analyzed thus far, it is possible that additional clades and different relationships will be identified as more populations are analyzed.
Since the present study was based on a limited number of nucleotide sites, future studies examining a greater proportion of nuclear genes may further define the population structure and genetic relationship between B. mori and B. mandarina from China and Japan. A better understanding of these features will increase our understanding of silkworm domestication and contribute to the phylogeographic studies of insect species.
ACKNOWLEDGMENT
We thank Nami Uwai and Naoko Kimura for providing technical support. We also thank Drs. Natuo Kômoto, Shuichiro Tomita, and two anonymous reviewers for their valuable comments and discussions.
This study was supported by a grant from the Ministry of Agriculture, Forestry and Fisheries, Research project for genomics for Agricultural Innovation GAM-111.
REFERENCES
- Aoki, K., Kato, M., and Murakami, N. (2008) Glacial bottleneck and postglacial recolonization of a seed parasitic weevil, Curculio hilgendorfi, inferred from mitochondrial DNA variation. Mol. Ecol. 17, 3276–3289.
- Clement, M., Posada, D., and Crandall, K. A. (2000) TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659.
- Guo, Y., Shen, Y. H., Sun, W., Kishino, H., Xiang, Z. H., and Zhang, Z. (2011) Nucleotide diversity and selection signature in the domesticated silkworm, Bombyx mori, and wild silkworm, Bombyx mandarina. J. Insect Sci. 11, 155.
- de Jong, M. A., Wahlberg, N., van Eijk, M., Brakefield, P. M., and Zwaan, B. J. (2011) Mitochondrial DNA signature for range-wide populations of Bicyclus anynana suggests a rapid expansion from recent refugia. PLoS ONE 6, e21385.
- Kawaguchi, E. (1928) Zytologische Untersuchungen am Seidenspinner und seinen Verwandten. I. Gametogenese von Bombyx mori L. und Bombyx mandarina M. und ihre Bastarde. Z. Zellforsch. Mikrosk. Anat. 7, 519–552.
- Kim, K. Y., Lee, E. M., Lee, I. H., Hong, M. Y., Kang, P. D., Choi, K. H., Gui, Z. Z., Jin, B. R., Hwang, J. S., Ryu, K. S., Han, Y. S., and Kim, I. (2008) Intronic sequences of the silkworm strains of Bombyx mori (Lepidoptera: Bombycidae): High variability and potential for strain identification. Eur. J. Entomol. 105, 73–80.
- Li, D., Guo, Y., Shao, H., Tellier, L. C., Wang, J., Xiang, Z., and Xia, Q. (2010) Genetic diversity, molecular phylogeny and selection evidence of the silkworm mitochondria implicated by complete resequencing of 41 genomes. BMC Evol. Biol. 10, 81.
- Librado, P., and Rozas, J. (2009) DnaSP v5: software for comprehensive analysis of DNA polymorphism. Bioinformatics 25, 1451–1452.
- McDonald, J. H., and Kreitman, M. (1991) Adaptive protein evolution at the Adh locus in Drosophila. Nature 351, 652–654.
- Nakamura, T., Banno, Y., Nakada, T., Nho, S. K., Xü, M. K., Ueda, K., Kawarabata, T., Kawaguchi, Y., and Koga, K. (1999) Geographic dimorphism of the wild silkworm, Bombyx mandarina in the chromosome number and the occurrence of a retroposon-like insertion in the arylphorin gene. Genome 42, 1117–1120.
- Osada, N., and Akashi, H. (2012) Mitochondrial- nuclear interactions and accelerated compensatory evolution: Evidence from the primate cytochrome c oxidase complex. Mol. Biol. Evol. 29, 337–346.
- Pan, M. H., Yu, Q. Y., Xia, Y. L., Dai, F. Y., Liu, Y. Q., Lu, C., Zhang, Z., and Xiang, Z. H. (2008) Characterization of mitochondrial genome of Chinese wild mulberry silkworm, Bomyx mandarina (Lepidoptera: Bombycidae). Sci. China Ser. C-Life Sci. 51, 693–701.
- Regier, J. C., Cook, C. P., Mitter, C., and Hussey, A. (2008) A phylogenetic study of the ‘bombycoid complex’ (Lepidoptera) using five protein-coding nuclear genes, with comments on the problem of macrolepidopteran phylogeny. Syst. Entomol. 33, 175–189.
- Tajima, F. (1989) The effect of change in population size on DNA polymorphism. Genetics 123, 597–601.
- Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011) MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739.
- Tanaka, H., Ishibashi, J., Fujita, K., Nakajima, Y., Sagisaka, A., Tomimoto, K., Suzuki, N., Yoshiyama, M., Kaneko, Y., Iwasaki, T., et al. (2008) A genome-wide analysis of genes and gene families involved in innate immunity of Bombyx mori. Insect Biochem. Mol. Bio. 38, 1087–1110.
- Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680.
- Tomita, S., Sohn, B. H., and Tamura, T. (1997) Cloning and characterization of a mariner-like element in the silkworm, Bombyx mori. Gen. Genet. Syst. 72, 219–228.
- Xia, Q., Guo, Y., Zhang, Z., Li, D., Xuan, Z., Li, Z., Dai, F., Li, Y., Cheng, D., Li, R., et al. (2009) Complete resequencing of 40 genomes reveals domestication events and genes in silkworm (Bombyx). Science 326, 433–436.
- Yu, H. S., Shen, Y. H., Yuan, G. X., Hu, Y. G., Xu, H. E., Xiang, Z. H., and Zhang, Z. (2011) Evidence of selection at melanin synthesis pathway loci during silkworm domestication. Mol. Biol. Evol. 28, 1785–1799.
- Yukuhiro, K., Sezutsu, H., Itoh, M., Shimizu, K., and Banno, Y. (2002) Significant levels of sequence divergence and gene rearrangements have occurred between the mitochondrial genomes of the wild mulberry silkmoth, Bombyx mandarina, and its close relative, the domesticated silkmoth, Bombyx mori. Mol. Biol. Evol. 19, 1385–1389.
- Yukuhiro, K., Sezutsu, H., Tamura, T., Kosegawa, E., and Kiuchi, M. (2011) Nucleotide sequence variation in mitochondrial COI gene among 147 silkworm (Bombyx mori) strains from Japanese, Chinese, European and moltinism classes. Genes Genet. Syst. 86, 315–323.