Special reviews
New horizons in genome engineering of Drosophila melanogaster
2014 Volume 89 Issue 1 Pages 3-8
Details
2014 Volume 89 Issue 1 Pages 3-8
Drosophila melanogaster has the longest history as a genetic model system and even in the present day remains the front runner in diverse fields of biology. However, lack of a convenient method to make specified modifications to endogenous genes has been a pain in the neck for many fly geneticists for decades. Synthetic nuclease technologies, especially the CRISPR/Cas9 system, hold great promise for a breakthrough. Synthetic nucleases are programmable nucleases that can be directed to cleave a specified sequence in the genome. Deleterious mutations can be efficiently induced by expression of a synthetic nuclease that targets a gene of interest. Precise modification of the target site, such as a reporter gene knock-in, is also possible by simultaneous delivery of a synthetic nuclease and a targeting vector. Here I summarize recent advances in synthetic nuclease technologies and discuss their possible applications to Drosophila genetics.
One of the major goals in modern biology is to completely understand how the genome works. The minimum requirement to achieve this goal would be to understand the function of each and every gene in the genome. Analysis of loss-of-function mutants of a given gene often provides a definitive insight to its biological function. Drosophila melanogaster has proven to be the most convenient multicellular organism for generating and analyzing mutants. Numerous forward genetic screens conducted over the past 100 years have identified thousands of mutants and their causative genes, thereby revealing core gene networks that govern diverse biological processes from early embryogenesis to complex adult behavior. Despite these efforts, more than 70% of the 14,000 Drosophila genes revealed by the complete genome sequence (Adams et al., 2000) have no described mutants and their physiological roles remain obscure. This highlights the fact that we are still far away from complete understanding of the genome even in this most extensively studied model organism. Thus, genetics in the post-genome era is in dire need of an efficient reverse genetics method to systematically disrupt all genes and interrogate their functions.
Reverse genetics in Drosophila has been conventionally done by imprecise excision of transposon insertions (Ryder and Russell, 2003). Targeted disruption of genes by homologous recombination (Rong and Golic, 2000), a standard technique in mouse genetics, has also been made possible in Drosophila. Both of these approaches, however, suffer from low efficiency and are not suitable for a systematic gene disruption. As an alternative to gene knock-out, gene knock-down by RNAi has been widely used for loss-of-function analysis in Drosophila. Genome-wide collections of transgenic RNAi lines are publicly available for screening (Leulier et al., 2002; Dietzl et al., 2007; Ni et al., 2011). Although RNAi screens have been useful for identifying new genes, target suppression is often insufficient to produce a discernable phenotype and it is difficult to estimate the false-negative rate. Therefore, genome-wide RNAi analysis also fails to provide a “complete” understanding of the genome. Recent advances in synthetic nuclease technologies are now changing the whole picture.
Synthetic nucleases are customizable nucleases that can be programmed to target any DNA sequence. They can be used to cut a unique site in the genome to induce mutations around the target site. The field was established by synthetic zinc-finger nucleases (ZFNs), heterodimeric nucleases in which each monomer comprises a nuclease domain derived from the FokI restriction enzyme and a synthetic DNA-binding polypeptide that comprise multiple zinc-finger domains that each recognize a specific 3-bp DNA sequence (Kim et al., 1996). Although many successful examples, including those in Drosophila, have been reported (Bibikova et al., 2002; Beumer et al., 2008), effective ZFNs were not easy to construct in part because zinc-finger units were not available for all of the 64 possible combinations of three base pairs and because simple concatenation of zinc-finger domains did not always result in desired target specificity. TALE nucleases (TALENs) are the second-generation synthetic nucleases and they fully address these problems. Like ZFNs, TALENs are built by fusing the FokI nuclease domain to a synthetic DNA-binding domain. Unlike ZFNs, the building blocks are DNA-binding peptides that recognize a single nucleotide, which can be simply concatenated to yield a polypeptide that binds a target sequence of any length (Miller et al., 2011). Because TALENs are relatively easy to construct and highly effective in a wide range of organisms including Drosophila (Liu et al., 2012; Katsuyama et al., 2013; Beumer et al., 2013; Kondo et al., 2014; Takasu et al., 2014), the research community welcomed TALENs with great enthusiasm and did not doubt that it was going to be the standard method of choice for animal genome engineering, until last year saw the advent of the CRISPR/Cas9 technology.
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) is an adaptive immunity system of bacteria in which the Cas9 nuclease cleaves the genomic DNA of incoming phages to prevent infection (Wiedenheft et al., 2012). The Cas9 nuclease is an RNA-guided endonuclease whose specificity is determined by the 20-bp sequence located at the 5′ end of its guide-RNA (gRNA) subunit (Fig. 1a). The Cas9-gRNA complex recognizes and cleaves a sequence in a double-strand DNA complementary to the 20-bp sequence of gRNA. Cas9 can be programmed to target virtually any sequence by simply changing the 20-bp gRNA sequence, the only constraint being that the 20-bp sequence must be followed by an NGG sequence known as the protospacer adjacent motif (PAM) (Jinek et al., 2012). Assuming that the base composition in the genome is homogeneous, such a sequence appears every 64 bp, making it possible to target almost all of the protein-coding genes. Because of its extremely easy design and construction, the CRISPR/Cas9 technology is rapidly replacing the other existing genome editing technologies. After the efficacy of Cas9 was first demonstrated in mammalian cells (Mali et al., 2013a; Cong et al., 2013), the last year has seen an avalanche of papers reporting genome modification in various animal species from Caenorhabditis elegans to monkeys (Wang et al., 2013; Friedland et al., 2013; Niu et al., 2014).
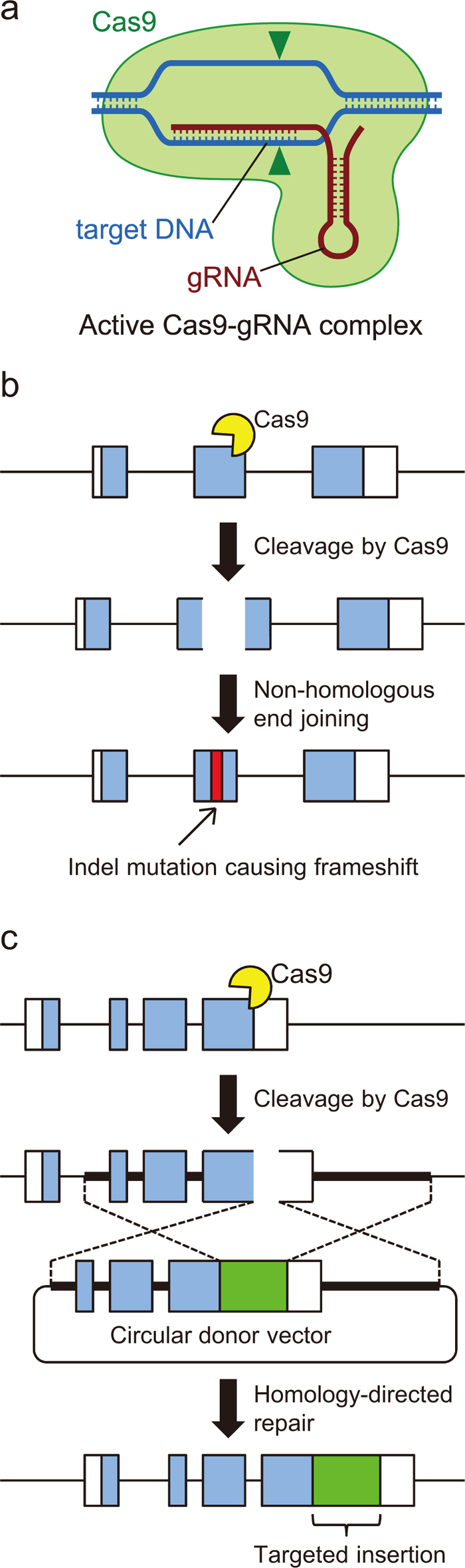
Strategies for genome engineering by CRISPR/Cas9. (a) Structure of the Cas9-gRNA complex. (b) Induction of indel mutations. When Cas9-induced DSBs are repaired by the NHEJ pathway, inaccurate repair results in indel mutations. (c) Targeted integration of a marker gene using a targeting donor vector. When homologous DNA is available nearby, DSBs are repaired through the HDR pathway using the homologous DNA as a template. The targeting vector has a marker gene (green box) flanked by homology arms (indicated by broken lines). HDR using the targeting vector results in seamless incorporation of the marker gene into the target site.
There are two major avenues through which synthetic nucleases are used for genome engineering. One is to induce random small insertions/deletions (indels) around the target site (Fig. 1b). Cleavage of DNA by synthetic nucleases leaves behind a double-strand break (DSB) at the target site. As DSBs are the most detrimental type of DNA damage, cells are equipped with multiple tiers of DNA repair systems to rapidly deal with DSBs (O’Driscoll and Jeggo, 2006). DSB repair is divided into two major classes: the precise homology-directed repair (HDR) pathway and the imprecise non-homologous end-joining (NHEJ) pathway. Although it is not fully understood how these two pathways are differentially selected, both pathways are active in animal cells including those of Drosophila (Johnson-Schlitz et al., 2007). Targeted induction of indels takes advantage of the NHEJ pathway. NHEJ is a simple form of DSB repair by which broken termini are brought together and joined by ligase. Since the broken termini are often processed to facilitate ligation, the repair outcome is typically inaccurate, with the sequence around the cleavage site harboring an indel of several base pairs (Yu and McVey, 2010). Thus, when synthetic nucleases are expressed in a cell, a certain fraction of the DSBs are repaired through the NHEJ pathway, resulting in indel mutations. In actual experiments, the final repair outcome is further biased toward inaccurate repair because overexpressed nuclease continues to cleave the target site as long as repair is accurate, until inaccurate repair renders the target site uncleavable due to sequence alteration. Indeed, the frequency of induced indels often approaches 100% using CRISPR/Cas9 (Wang et al., 2013; Kondo and Ueda, 2013).
The other major application of synthetic nucleases is precise modification of target loci, or gene knock-in, using a donor template (Fig. 1c). Unlike induction of random indels, precise target modification takes advantage of the HDR pathway of DSB repair. Under natural conditions, DSB repair by HDR involves the copying of a sequence of the sister chromatid or the homologous chromosome to fill the gap between the broken termini and restore the original sequence (Adams et al., 2003). In genome engineering, a synthetic nuclease and a donor DNA vector with homology to the sequence around the cleavage are simultaneously delivered into cells, tricking the cells into using the donor vector as a repair template and incorporating a modified allele carried in the vector. The donor vector can have various types of modifications including single-nucleotide changes, small indels, kilobase-sized deletions and insertion of large exogenous DNA fragments such as selection markers.
Implementation of the CRISPR/Cas9 technology in Drosophila for genome engineering has been independently reported by several groups (Gratz et al., 2013; Bassett et al., 2013; Yu et al., 2013; Kondo and Ueda, 2013; Sebo et al., 2013; Ren et al., 2013). In these studies, Cas9-gRNA was expressed in germ cells to induce heritable indel mutations. They differ in the way Cas9-gRNA is delivered into germ cells. Gratz et al. (2013) were the first to report successful gene disruption by CRISPR/Cas9. They co-injected two plasmid expression vectors, each encoding Cas9 protein and gRNA, into fertilized eggs. They observed an average mutation frequency of 1% in the progeny of the injected parents, which was rather low from a practical point of view. The low efficiency may have been due to the poor activity of the heat-shock promoter used to drive the expression of Cas9, whereas injection of TALEN-expressing plasmids, in which a copia promoter is used, is able to induce mutations with efficiency higher by an order of magnitude (Katsuyama et al., 2013). Subsequent studies showed that mutagenesis efficiency could be significantly improved by co-injection of in vitro transcribed gRNA and mRNA encoding Cas9, with an average mutation frequency of 20–30% (Bassett et al., 2013; Yu et al., 2013). Potential disadvantages of this approach are that the prepared RNA must be handled with extreme care due to its vulnerability to RNase and that the required reagents are not inexpensive. To provide a more convenient source of Cas9, we and others constructed transgenic lines that express Cas9 specifically in germ cells from the germline-specific nos or vasa promoter (Kondo and Ueda, 2013; Sebo et al., 2013; Ren et al., 2013). Injection of gRNA expression vectors, which express gRNA from the ubiquitous U6-promoter, into Cas9-expressing eggs induced mutations with efficiency comparable to that by injection of Cas9 mRNA and gRNA (Sebo et al., 2013; Ren et al., 2013; Kondo, unpublished observation). We attempted to further improve the mutagenesis efficiency by also providing gRNA as a transgene. We established transgenic lines expressing gRNA from a U6 promoter. By crossing nos-Cas9 with U6-gRNA lines, we could induce targeted mutations in the germline of F1 progeny at frequencies often exceeding 80% (Kondo and Ueda, 2013). Thus, screening of a mere eight mutant candidates usually yields multiple null alleles. In addition, our all-transgenic approach also allows researchers to free themselves from time-consuming injection experiments by using external injection services to construct gRNA transgenic flies.
CRISPR/Cas9 can also be used for precise genome modification using targeting vectors. Baena-Lopez et al. (2013) reported successful knock-in of a large replaceable cassette albeit at the low frequency of 0.2%. Again, the efficiency could be dramatically improved by using a transgenic source of Cas9. Gratz et al. (2014) reported a targeting efficiency of more than 10% (transgenic founder/injected parents) by injecting a mixture of a targeting vector and a gRNA expression vector into vas-Cas9 eggs. We have obtained similar efficiency using our nos-Cas9 strain (Kondo, unpublished observation). In conventional gene targeting that does not use synthetic nucleases, successful targeting required homology arms longer than 5 kb in the targeting vector. In contrast, if the target site is cleaved by synthetic nuclease, 0.5-kb homology arms are sufficient to achieve maximal efficiency (Urnov et al., 2005; Beumer et al., 2008, 2013). The smaller size allows for PCR-amplification of homology arms, dramatically facilitating vector construction. Another point to note is that, unlike conventional gene targeting by homologous recombination, circular donor vectors are far more efficient than linearized donor vector, most likely because linear vectors are rapidly degraded by endogenous exonuclease (Beumer et al., 2008, 2013).
Off-target cleavage is a major concern for synthetic nuclease technologies. Both in vivo and in vitro, several studies have shown that Cas9 is capable of cleaving a target site with up to five mismatches albeit with reduced efficiency (Hsu et al., 2013; Mali et al., 2013b; Pattanayak et al., 2013). Although no cases of off-target cleavage have been confirmed in Drosophila, it would be best to avoid target sequences with potential off-targets. Several webtools are available to facilitate identification of highly specific targets (Ren et al., 2013; Gratz et al., 2014).
As I have discussed, targeted gene disruption is now extremely easy using CRISPR/Cas9, to the point where it is realistic to construct a mutant library covering all of the 14,000 protein-coding genes in the Drosophila genome. With the goal of producing a genome-scale mutant collection, we have optimized our pipeline for generating mutants by CRISPR/Cas9 (Fig. 2). Currently, we have the capacity to generate mutants in 50–100 genes per month at the cost of $50 per gene with an average turnaround time of 2 months. By combining efforts of multiple laboratories, it would be possible to generate all of the mutants in a few years’ time.

Crossing scheme for generating Cas9-induced mutants. A mutagenesis method for third chromosome genes is shown. In Step 1, a gRNA-expression plasmid (U6-gRNA) is injected into the nos-phiC31; attP2 host for targeted integration into the attP2 landing site. The injected eggs are raised to adulthood. Males are crossed to balancer females (y w; Dr/TM6B) in Step 2. We set up five crosses, of which two or three usually produce transgenic offspring. Since U6-gRNA is marked with a vermilion transgene, recombinant flies can be identified as vermilion+ flies. In Step 3, transgenic males of the y w; U6-gRNA/TM6B genotype are picked and crossed to y w; nos-Cas9/CyO females. Among their progeny, male flies carrying nos-Cas9 and U6-gRNA are crossed to balancer females in Step 4. In Step 5, male mutant candidates that carry neither of the transgenes are selected and crossed to balancer females. Three days after mating, male flies are removed from the vials and individually subjected to PCR and sequence analysis. If a mutation is detected, progeny derived from the mutation carrier are used to establish a stock. The overall procedure is essentially the same for second chromosome genes, except that y w; Sp/CyO is substituted for y w; Dr/TM6B in Steps 4 and 5.
Precise modification of endogenous genes will also open the door to novel genetic experiments that have hitherto been impossible or extremely difficult. It will become possible to address the function of a particular domain or a particular amino acid of a protein in an endogenous context by swapping domains or substituting particular amino acids in endogenous genes. Insertion of reporter genes, such as GAL4 and LexA, into the translation initiation site of a gene will allow monitoring of gene expression with better accuracy than enhancer traps. In-frame tagging of genes with fluorescent proteins will allow analysis of in vivo protein dynamics at the endogenous level of expression.
In the present day, Drosophila is actively used as a key model system in a wide range of biological fields including such intensely studied subjects as cancer biology, stem cell biology, developmental biology and neurobiology (for specific examples, see Niwa and Niwa, 2014, Sakuma et al., 2014, Hakeda-Suzuki and Suzuki, 2014). The new resources and methods brought about by the synthetic nuclease technology will no doubt contribute to systems-level understanding of the complex molecular mechanisms underlying these important biological processes.