Full papers
AbpA and AbpB provide anti-phage activity in Escherichia coli
2014 Volume 89 Issue 2 Pages 51-60
Details
2014 Volume 89 Issue 2 Pages 51-60
Bacteria have a variety of resistance mechanisms for surviving bacteriophage infections. Here, we describe a novel anti-phage mechanism in Escherichia coli. Cells harboring a plasmid with the genes abpA and abpB, formerly yfjL and yfjK, blocked the propagation of bacteriophages belonging to three families: T4, T2, T7 and λ phages. Both genes were necessary for the inhibition of phage propagation, and deletion of either chromosomal gene resulted in a 20% increase of progeny compared to wild-type cells. Neither overexpression nor deficiency of AbpA and AbpB had any apparent effect on E. coli growth. We isolated seven suppressor mutants of T4 phage that grew weakly on cells overexpressing AbpA and AbpB, and found that their mutations were all located in gene 41, which encodes a replicative DNA helicase that is essential for DNA replication. Furthermore, we demonstrated that AbpA and AbpB inhibited DNA replication and late gene expression of T4 phage. Similarly, DNA replication of T7 and λ phages was also inhibited by AbpA and AbpB. These results strongly suggest that E. coli AbpA and AbpB target DNA replication of phages to block their propagation.
Bacteriophages are viruses that infect bacteria and are the most abundant biological entities (Bergh et al., 1989; Wommack and Colwell, 2000; Srinivasiah et al., 2008). Since phages outnumber bacteria by approximately 10-fold (Brüssow and Hendrix, 2002), bacteria are constantly exposed to invasion by phages (Suttle, 2007). To combat this threat, bacteria have evolved a variety of resistance mechanisms (Sturino and Klaenhammer, 2006; Labrie et al., 2010; Bikard and Marraffini, 2012). On the other hand, phages have also evolved mechanisms to overcome these bacterial anti-phage mechanisms (Otsuka and Yonesaki, 2012; Samson et al., 2013). Accordingly, phages and bacteria co-evolve (Comeau and Krisch, 2005; Kashiwagi and Yomo, 2011; Stern and Sorek, 2011).
Bacterial anti-phage mechanisms target all stages of phage replication (Labrie et al., 2010). For example, bacteria have changed their outer surfaces to evade phage adsorption (Tamaki et al., 1971; Riede and Eschbach, 1986; Reyes-Cortés et al., 2012), or have developed restriction enzymes (Bickle and Krüger, 1993; Tock and Dryden, 2005) and CRISPR systems (Barrangou et al., 2007; Marraffini and Sontheimer, 2010) to destroy invading DNA. Additionally, bacteria carry many abortive infection (Abi) systems that rapidly induce the death of phage-infected bacteria to prevent phage transmission to uninfected cells. The Rex system is the best characterized Abi system found in phage λ-lysogenic Escherichia coli strains and is composed of two proteins, RexA and RexB (Parma et al., 1992; Snyder, 1995). RexA senses phage infection by producing a phage protein-DNA complex, and then activates RexB. RexB is an ion channel protein whose activation leads to a loss of membrane potential and a reduction of the ATP level in the cell, and consequently stops bacterial growth and the production of phage progeny. Intriguingly, at least 23 distinct Abi systems have been reported in Lactococcus lactis (Chopin et al., 2005). They affect many different stages of phage replication cycle, such as DNA replication, transcription, mRNA stability, capsid protein production, phage DNA packaging and lysis (Labrie et al., 2010), but the mechanistic details of these systems remain unclear. Some bacterial toxin-antitoxin (TA) systems also have been reported to play a role in phage defense (Pecota and Wood, 1996; Hazan and Engelberg-Kulka, 2004; Fineran et al., 2009; Samson et al., 2013; Sberro et al., 2013). TA systems are broadly conserved in plasmids and bacterial chromosomes (Gerdes et al., 2005), and are implicated in many physiological roles, such as plasmid maintenance (Ogura and Hiraga, 1983; Yarmolinsky, 1995), stress response (Gerdes et al., 2005; Wang et al., 2011), bacterial persistence against antibiotics (Maisonneuve et al., 2011, 2013; Amato et al., 2013; Germain et al., 2013) and biofilm formation (Kim et al., 2009; Wang et al., 2011). We have demonstrated that the two toxins RnlA and LsoA in TA systems are activated after T4 phage infection and result in shutting off gene expression by rapidly degrading mRNAs to block T4 propagation (Kai et al., 1996; Otsuka and Yonesaki, 2005, 2012; Koga et al., 2011).
In this study, we discovered a new anti-phage mechanism in E. coli. Cells harboring a plasmid with the previously uncharacterized genes yfjL and yfjK almost completely blocked the propagation of representatives of different families of phages: T4, T2, T7 and λ. Based on this observation, we renamed yfjL and yfjK as abpA (anti-bacteriophage protein A) and abpB (anti-bacteriophage protein B), respectively. The two genes form an operon and are located in the region of prophage CP4-57 (Wang et al., 2009). The coding sequences of abpA and abpB overlap by 4 bp, which may facilitate the coupling of their expression at the translational level, as has been described for other proteins (Normark et al., 1983). AbpA (538 aa) and AbpB (729 aa) are broadly conserved in bacteria and form an operon in many, but not all, cases. No conserved domain was detected in AbpA, but AbpB possesses a DEAD-box helicase domain, which is involved in various aspects of RNA metabolism (Kaberdin and Bläsi, 2013). As an initial step for understanding this broad anti-phage mechanism, we addressed the target of AbpA and AbpB in the inhibition of phage growth.
E. coli K-12 strains MH1 (sup0 araD139 hsdR ΔlacX74 rpsL) and TY0807 (MH1 araD+) were used as wild types (Koga et al., 2011). TY0832 (MH1 ΔabpA::kan) and TY0833 (MH1 ΔabpB::kan) were constructed by GT7-mediated transduction of a kanamycin-resistance marker from strains JW2609 (BW25113 ΔyfjL::kan) and JW2608 (BW25113 ΔyfjK::kan), which were provided by the National BioResource Project (NIG, Japan). TY0836 (MH1 ΔabpA-abpB) and TY0837 (TY0807 ΔabpA-abpB) were constructed as described (Datsenko and Wanner, 2000). Briefly, a fragment containing a chloramphenicol-resistance cassette flanked with the sequences upstream of abpA and downstream of abpB was amplified by PCR with pKD3 as a template and the primers 5’-ACCTGCGACCGAGAATGGCTGTGAAGTATTTAGATCACTGGTGATGATGCATGGGAATTAGCCATGGTCC and 5’-GTGAGTTATTCAAAAATGTCGCTTTCCCGCGTTCCGTAGACAAACGTACCGTGTAGGCTGGAGCTGCTTC. The fragment was introduced into MH1 or TY0807 harboring pKD46, which encodes the λ phage Red recombinase, and chloramphenicol-resistant colonies were screened by PCR with the primers 5’-ACCTGCGACCGAGAATGGCTGTG and 5’-GTGAGTTATTCAAAAATGTCGC to select TY0834 (MH1 ΔabpA-abpB::cat) or TY0835 (TY0807 ΔabpA-abpB::cat). The chloramphenicol-resistance cassette was removed by yeast Flp recombinase expressed from pCP20 (Cherepanov and Wackernagel, 1995) to construct TY0836 or TY0837. Bacteriophages were T4D, T2L, T7 and λ RS45(bla’-lacZSC) (Simons et al., 1987).
Construction of plasmidspST4-1 contains a 4512-bp DNA fragment (2760763-2765275 of GenBank Accession No. NC_000913.3). To construct pBAD33-abpA, a DNA fragment was amplified by PCR using pST4-1 as a template and the primers 5’-TTTGTCTAGAAAATTGTCACGGATGTCTAAATG and 5’-TTTCAAGCTTTCTGTCATACGACCTCTCTCAC, digested with NheI and HindIII, and cloned into XbaI- and HindIII-digested pBAD33 (Guzman et al., 1995). To generate pQE80L-abpB, a DNA fragment was amplified by PCR using pST4-1 as a template and the primers 5’-GGAATTCAGAAGGTGAGAGAGGTCGTATG and 5’-CGGATCCTAAATCAATGTCGTCTAATACC, digested with EcoRI and BamHI, and ligated into the corresponding sites of pQE80L (QIAGEN). To construct pBAD24-abpA-Flag, a DNA fragment was amplified by PCR using pST4-1 as a template and the primers 5’-AAAAACCATGGAGTCGAACGTTCAGGCG and 5’-GTCTAAGCTTCACTTGTCATCGTCGTCCTTGTAGTCTACGACCTCTCTCACCTTCTC. The DNA fragment was digested with NcoI and HindIII, and ligated into the corresponding sites of pBAD24 (Guzman et al., 1995). pBAD24-abpA-Flag was digested with BamHI and HindIII and ligated into pBAD33 to construct pBAD33-abpA-Flag. To generate pQE80L-His-abpB, a DNA fragment was amplified by PCR using pST4-1 as a template and the primers 5’-AAAGGATCCACAGAGATCTATGAACAGGC and 5’-TCAAGCTTTCAATGTCGTCTAATACCGAG. The DNA fragment was digested with BamHI and HindIII, and ligated into the corresponding sites of pQE80L.
Pull-down assay and Western blot analysisTY0807 cells harboring pBAD33-abpA-Flag plus either pQE80L-His-abpB or pQE80L-His-Rng encoding His-tagged Rng (Koga et al., 2011) were grown at 30℃ in 15 ml of LB medium containing 50 μg ml–1 ampicillin and 30 μg ml–1 chloramphenicol and, when the OD600 reached 0.3, AbpA-FLAG and His-AbpB were co-induced with 0.2% L-arabinose and 0.1 mM IPTG for 1 h. Cells were harvested and suspended in 1.5 ml of lysis buffer [10 mM Tris-HCl (pH 7.5), 30 mM KCl, 10 mM magnesium acetate, 20 mM imidazole]. After sonication, the lysate was centrifuged at 9,000 g for 20 min and the protein concentration of the supernatant was determined by Bradford protein assay. Cell extract (400 μg) was mixed with 20 μl of Ni-NTA Superflow agarose beads (QIAGEN) by end-over-end rotation overnight at 4℃. The beads were washed five times with 1 ml of washing buffer [10 mM Tris-HCl (pH 7.5), 10 mM magnesium acetate, 100 mM KCl, 0.5 mM DTT, 20 mM imidazole]. Bound proteins were eluted with 70 μl of elution buffer [10 mM Tris-HCl (pH 7.5), 10 mM magnesium acetate, 100 mM KCl, 0.5 mM DTT, 400 mM imidazole]. Proteins were separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and electroblotted onto Immuno-Blot PVDF membrane (Bio-Rad). The membranes were probed with a mouse anti-FLAG M2 monoclonal antibody (Sigma-Aldrich) or a mouse anti-His antibody (GE Healthcare) and then with horseradish peroxidase-conjugated sheep anti-mouse IgG (GE Healthcare). Protein bands were detected with Immobilon Western Chemiluminescent Substrate (Millipore) and an LAS image analyzer (Fujifilm).
Labeling of newly synthesized proteinsCells were grown to a density of 5 × 108 cells ml–1 at 30℃ in M9 minimal medium supplemented with 0.3% casamino acids, 1 μg ml–1 thiamine and 20 μg ml–1 tryptophan, and infected with T4 phage at a multiplicity of infection (m.o.i.) of 7. [35S]methionine/cysteine (American Radiolabeled Chemicals; >37 Tbq mmol–1) was added at 3.7 MBq ml–1 to 0.1-ml cultures at various times after infection. After incubation for 3 min, incorporation of radioactive amino acids was stopped by adding 2 μl of 20% casamino acids and chilling the tubes quickly on ice. Cells were centrifuged and suspended with sample loading buffer. After boiling, labeled proteins were separated by 12.5% SDS-PAGE and analyzed using a Bio-Image Analyzer (Fuji BAS-1800).
RNA purification and Northern blot analysisThese were carried out as described (Kai et al., 1996). The radioactive probes for gene 23 and 24 transcripts were prepared as described (Ueno and Yonesaki, 2001).
Southern dot blot analysisMH1 cells harboring pBR322 or pST4-1 were grown in M9 minimal medium supplemented with 0.3% casamino acids, 1 μg ml–1 thiamine and 20 μg ml–1 tryptophan until the OD600 reached 0.5, and infected with T4 or T7 phage at an m.o.i. of 5. In the case of λ phage, λ broth was used and the cells were infected at an m.o.i. of 1 when the OD600 reached 0.4. Aliquots (100 μl) of cell cultures were collected at 0, 10, 20, 30, 40 and 50 min after infection for T4 and T7 phages and at 0, 30, 60, 90, 120, 150 and 180 min for λ phage. After centrifugation, the cell pellets were suspended in 20 μl (T4 phage) or 10 μl (T7 or λ phage) of phosphate buffer, and then 3 μl (T4 or T7 phage) or 5 μl (λ phage) of each cell suspension was spotted onto a Hybond N+ nylon membrane (GE Healthcare). The membrane was treated sequentially with 10% SDS for 3 min, buffer B [0.5 N NaOH and 1.5 M NaCl] for 5 min, buffer C [0.5 M Tris-HCl (pH7.5) and 1.5 M NaCl] for 5 min, and 1× SSC for 5 min. After fixing the nucleic acid to the membrane by UV irradiation, the membrane was hybridized with labeled probe at 42℃ overnight. Radioactive probes for T4, T7 and λ phage DNA were prepared by PCR using one primer 5’ end-labeled with T4 kinase and [γ-32P]ATP, another unlabeled primer and phage genomic DNA as a template. Primers used for PCR were as follows: for T4 phage, 5’-32P-CCTGCAGTAACAAGTTCGGCTC and 5’-CCTGCAGTACTCTCCTCTATA; for T7 phage, 5’-32P-AGGAATTCGATATTCACTAATAACTGCACGAGG and 5’-CGCTAAAGATGTCAGCGTAG; and for λ phage, 5’-32P-ATGGATCAAACACTTATGGC and 5’-TTAGTGAATGCTTTTGCTTG.
Previously we identified rnlA in E. coli as a gene responsible for blocking T4 phage propagation (Otsuka and Yonesaki, 2005). During that process, we constructed a shotgun library, in which DNA fragments obtained from digestion of MH1 DNA with HindIII were cloned in pBR322, and found that one clone did not support the growth of T4 phage. The plasmid, named pST4-1, recovered from this clone was sequenced and found to contain two complete ORFs of genes of unknown function, yfjL and yfjK. To investigate the effect of pST4-1 on phage growth, T4, T2, T7 and λ phages were spotted on plates seeded with MH1 cells containing pBR322 or pST4-1, and incubated at 30℃ overnight (Fig. 1). Every phage grew normally on cells containing pBR322, but their efficiencies of plating decreased dramatically to less than 10–4 in the presence of pST4-1. This inhibitory effect of pST4-1 on T4 phage growth was temperature-dependent, and was barely detectable at 37℃ or 42℃ (Supplementary Fig. S1). We also confirmed the effect of pST4-1 on propagation of T4 phage by measuring burst size (Table 1). T4 phage grew very poorly on cells harboring pST4-1 and its burst size was only 0.1, while it grew well on cells harboring pBR322, with a burst size of 53.
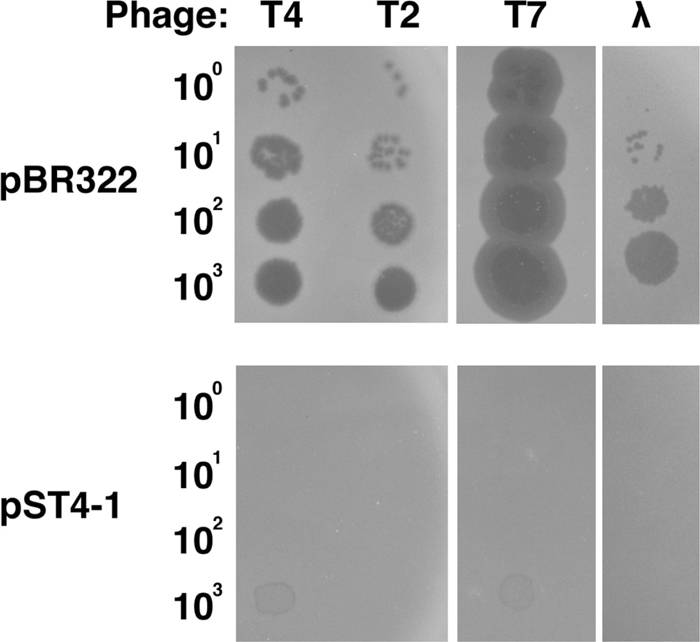
Effect of pST4-1 on phage growth. Suspensions containing the number of T4, T2, T7, and λ phage particles indicated on the left were spotted onto plates seeded with MH1 cells harboring pBR322 or pST4-1 and the plates were incubated at 30℃ overnight.
| Plasmid | Burst size |
|---|---|
| pBR322 | 53.1 ± 6.6 |
| pST4-1 | 0.1 ± 0.02 |
MH1 cells harboring pBR322 or pST4-1 were grown in LB medium until the OD600 reached 0.4, and infected with T4 phage at an m.o.i. of 0.1 at 30℃. At 8 min, the cells were diluted 104-fold with fresh LB and further incubated for 70 min. After the cells were lysed with chloroform, the total number of progeny phage particles was determined by plating. The burst size is the ratio of the number of progeny to the number of input phage particles. Each value is the mean and standard deviation of at least three independent experiments.
Next, we tried to identify which gene, abpA or abpB, is necessary for inhibition of T4 phage propagation. For this purpose, we cloned a DNA fragment encoding AbpA into pBAD33 (pBAD33-abpA) under the control of an arabinose-inducible promoter, and another encoding AbpB into pQE80L (pQE80L-abpB) under the control of an IPTG-inducible promoter. TY0837 (ΔabpA-abpB) cells containing pBAD33 or pBAD33-abpA were co-transformed with pQE80L or pQE80L-abpB. These cells were grown to mid-log phase, treated with arabinose and IPTG for 45 min, and then infected with T4 phage. When only AbpA or AbpB was expressed, T4 phage grew well with a burst size comparable to that on cells harboring pBAD33 and pQE80L (Table 2). However, the burst size on cells co-expressing AbpA and AbpB was very low, and similar to that on cells carrying pST4-1. These data indicated that both AbpA and AbpB were required for the inhibition of T4 propagation. To investigate the effect of endogenous AbpA and AbpB on phage growth, we measured burst sizes of T4 phage on MH1 (wild type), TY0832 (ΔabpA::kan), TY0833 (ΔabpB::kan) or TY0836 (ΔabpA-abpB) cells (Table 3). The effects of the endogenous genes were modest; burst sizes were increased 1.2-fold by disruption of abpA, abpB, or both, compared with the burst size on wild type cells. This result also demonstrates that both abpA and abpB are necessary for inhibition of phage propagation.
| Plasmid | Burst size |
|---|---|
| pBAD33 + pQE80L | 39.5 ± 1.8 |
| pBAD33-abpA + pQE80L | 43.5 ± 5.8 |
| pBAD33 + pQE80L-abpB | 45.2 ± 8.4 |
| pBAD33-abpA + pQE80L-abpB | 0.3 ± 0.1 |
TY0837 (ΔabpA-abpB) cells harboring pBAD33 or pBAD33-abpA, plus pQE80L or pQE80L-abpB, were grown in LB medium until the OD600 reached 0.3, then treated with 0.2% L-arabinose and 50 μM IPTG for 45 min, and infected with T4 phage at an m.o.i. of 0.1 at 30℃. The burst size was determined as described in Table 1.
| Genotype | Burst size |
|---|---|
| Wild type | 60.1 ± 2.7 |
| ΔabpA | 71.6 ± 6.8 |
| ΔabpB | 75.0 ± 3.5 |
| ΔabpA-abpB | 73.7 ± 4.9 |
MH1 (wild type), TY0832 (ΔabpA::kan), TY0833 (ΔabpB::kan) and TY0836 (ΔabpA-abpB) cells were grown in PB medium [1% casein-peptone, 0.1% glucose, 0.3% NaCl, 100 μM CaCl2, 1 mM MgCl2, 320 μM KH2PO4 (pH 7.0)] until the OD600 reached 0.4, and then infected with T4 phage at an m.o.i. of 0.1 at 30℃. At 8 min, the cells were diluted 104-fold with fresh PB and further incubated for 70 min. The burst size was determined as described in Table 1.
The necessity of two genes for inhibition of T4 propagation suggested that AbpA and AbpB interact with each other. Hence, we carried out a pull-down experiment to examine their interaction in vivo. We constructed two plasmids, pBAD33-abpA-Flag expressing FLAG-tagged AbpA and pQE80L-His-abpB expressing His-tagged AbpB. Cell extracts were prepared from TY0837 harboring pBAD33-abpA-Flag plus either pQE80L-His-abpB or pQE80L-His-Rng as a negative control. Rng is an endoribonuclease G which is involved in the processing of the 5’ end of 16S rRNAs and the degradation of some mRNAs (Tock et al., 2000). After mixing cell extracts with Ni-NTA beads and then washing with buffer, bound proteins were analyzed by Western blotting with an antibody against FLAG-tag or His-tag (Fig. 2). AbpA-FLAG was efficiently recovered in the bound fraction together with His-AbpB, but was undetectable in the bound fraction with His-Rng, indicating that AbpA and AbpB interact with each other in vivo.

Interaction between AbpA and AbpB in vivo. Extracts of TY0837 cells harboring pBAD33-abpA-Flag plus either pQE80L-His-abpB or pQE80L-His-Rng (control) were subjected to pull-down with Ni-NTA agarose beads. Input and bound fractions were analyzed by Western blot with anti-FLAG or anti-His antibody as described in MATERIALS AND METHODS.
To investigate why phage growth is blocked in the presence of AbpA and AbpB, we investigated their effect on T4 gene expression. Gene expression at each stage after infection was monitored by pulse-labeling newly synthesized proteins (Fig. 3a). In MH1 cells harboring pBR322, different classes of proteins synthesized sequentially were discernible as reported (Kai et al., 1996) for the representative pattern of T4 gene expression: early (3–6 min after T4 infection), middle (10–13 min) and late (23–26, 40–43 and 80–83 min) proteins. In cells harboring pST4-1, late proteins were hardly synthesized and middle proteins were still being expressed at 23, 40 and 80 min, while early and middle proteins were synthesized with similar kinetics and abundance to those in cells harboring pBR322.
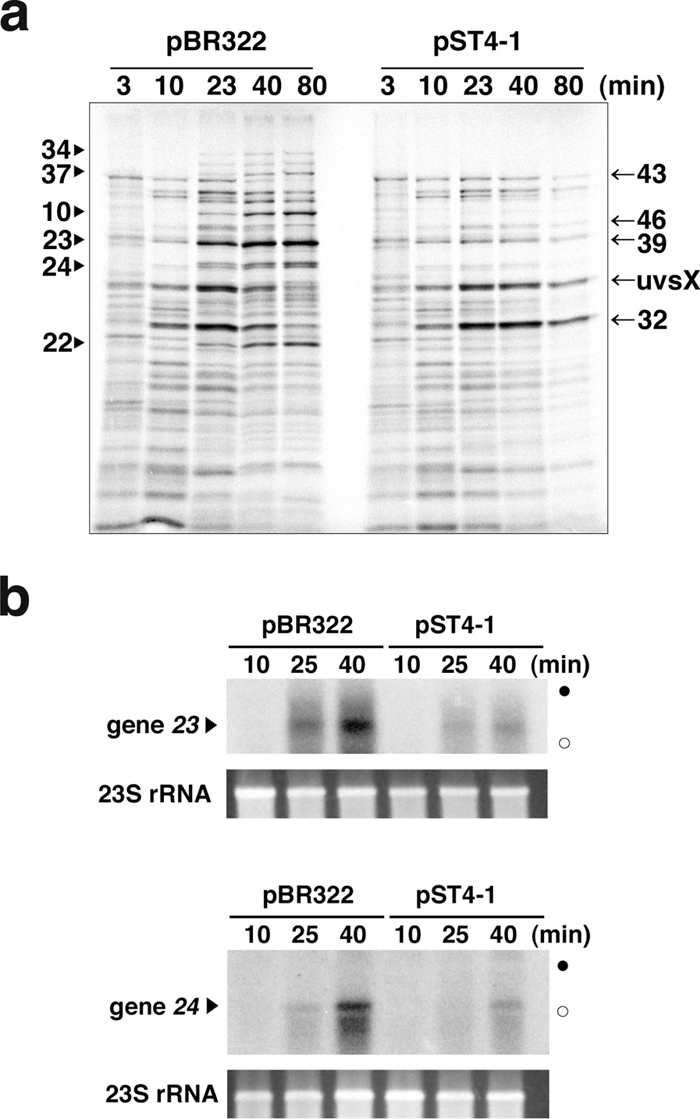
Effect of AbpA and AbpB on gene expression of T4 phage. (a) MH1 cells harboring pBR322 or pST4-1 were infected with T4 phage and newly synthesized proteins were pulse-labeled for 3 min, at the times indicated above the image, as described in MATERIALS AND METHODS. Labeled proteins from equal numbers of infected cells were separated by 12.5% SDS-PAGE and detected with a Bio-Image Analyzer. Representative late (arrowheads) and middle T4 proteins (arrows) are indicated at the left and right margins, respectively. (b) MH1 cells harboring pBR322 or pST4-1 were infected with T4 phage at time 0 as described in MATERIALS AND METHODS. Total RNAs were extracted at 10, 25 and 40 min, and 5 μg of each RNA was electrophoresed through a polyacrylamide gel and analyzed by Northern blotting against gene 23 (upper panel) and 24 (lower panel). Closed and open circles mark the positions of 23S and 16S rRNAs. Ethidium bromide-stained 23S rRNAs are shown at the bottom of each panel.
We next checked the expression of late-gene transcripts by Northern blotting (Fig. 3b). As expected, the amounts of gene 23 and 24 transcripts in the presence of pST4-1 were markedly lower, at 25 and 40 min after T4 infection, than those in the presence of pBR322. Taken together, these data indicate that AbpA and AbpB impaired the synthesis of late gene transcripts, which resulted in poor expression of late proteins and consequently no phage propagation.
Isolation of T4 mutants that can form plaques in the presence of pST4-1The transcription of T4 late genes depends on two different molecular events: a switch of the sigma factor to one encoded by gene 55, and ongoing DNA replication (Geiduschek and Kassavetis, 2010). To investigate which is hampered by AbpA and AbpB, we isolated 11 spontaneous T4 mutants that were able to grow in the presence of pST4-1. These mutants formed tiny but clear plaques on cells harboring pST4-1 under conditions in which wild type phage could not form a plaque at all (Fig. 4, middle panel).
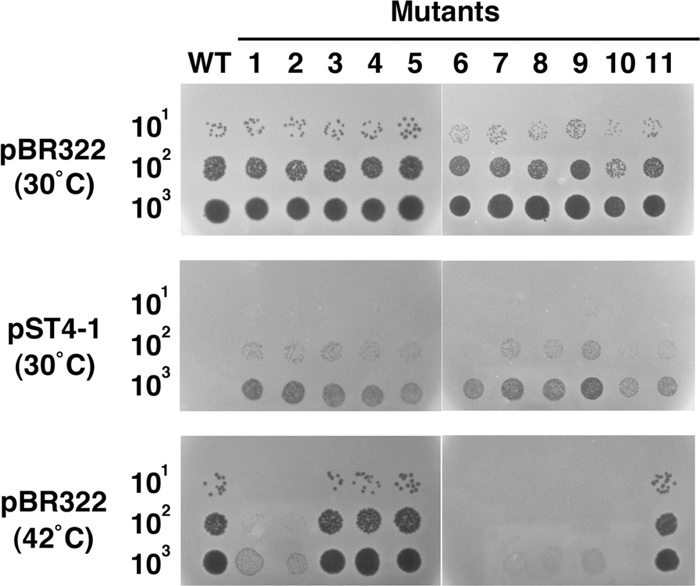
Effect of suppressor mutations on the growth of T4 phage. Suspensions containing the number of wild type or suppressor mutant T4 phage particles indicated on the left were spotted onto plates seeded with MH1 cells harboring pBR322 or pST4-1, and the plates were incubated at 30℃ or 42℃ overnight.
Among these 11 suppressor mutants, seven could not grow on cells harboring pBR322 at 42℃ (Fig. 4, bottom panel), suggesting that they have a temperature-sensitive (ts) mutation in an essential gene. The recombination frequency between them and representative genetic markers indicated that all seven ts mutations resided in the neighborhood of gene 41. Furthermore, the low recombination frequency (less than 0.1%) observed in crosses with a mutant of gene 41 suggested that these ts mutations were located within gene 41. To test this, we examined whether or not the ts phenotypes were suppressed by plasmid-borne gene 41. All seven mutants could grow at 42℃ on MH1 cells harboring p415, which carries gene 41 (Yonesaki, 1994; data not shown). Finally, sequencing revealed that six of the seven ts mutants had an identical one-base substitution in gene 41: G at nucleotide 637 within the coding region was changed to T, replacing Asp at codon 213 with Tyr. The seventh ts mutant also had an one-base substitution in gene 41: T at nucleotide 575 was changed to A, replacing Leu at codon 192 with Gln.
To confirm that the ts mutations in gene 41 render T4 phage able to grow on MH1 cells harboring pST4-1, the mutations were marker-rescued by homologous recombination between genomic DNA of ts suppressor mutant phages and p415. The resulting temperature-resistant phages were examined for growth ability on cells harboring pST4-1 at 30℃, and were found to be unable to grow (Supplementary Fig. S2). This result indicates that ts mutations in gene 41 correspond to suppressor mutations to support the growth of T4 phage in the presence of pST4-1.
Effect of AbpA and AbpB on phage DNA replicationT4 gene 41 encodes a replicative DNA helicase that is a subunit of the primosome, which is essential for lagging-strand DNA synthesis (Jones et al., 2001). Because at least seven out of 11 suppressor mutants have a mutation in gene 41, and because the defect in T4 late gene expression in the presence of pST4-1 may result from impaired DNA replication (see DISCUSSION), we hypothesized that the cause of abortive T4 propagation might be that AbpA and AbpB impaired phage DNA replication. To examine this possibility, we measured the amount of DNA synthesized after infection with T4, T7 and λ phages by Southern dot blotting (Fig. 5). In cells harboring pBR322, the amount of phage DNA increased with time after infection. On the other hand, no DNA synthesis of any phages was evident in the presence of pST4-1. This result clearly showed that AbpA and AbpB impair the synthesis of phage DNA.
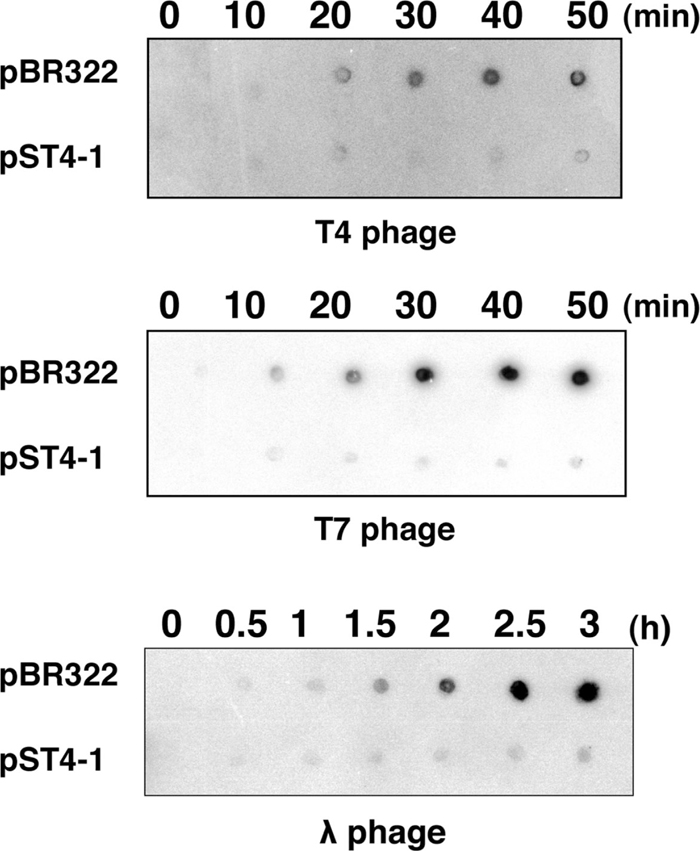
Effect of AbpA and AbpB on phage DNA synthesis. MH1 cells harboring pBR322 or pST4-1 were grown and infected with T4 (top), T7 (middle) or λ (bottom) phages. Total DNA was extracted at the indicated times after infection and analyzed by Southern dot blotting as described in MATERIALS AND METHODSs.
To investigate whether the effect of AbpA and AbpB is specific to phage growth, we compared bacterial growth rates in the presence of overexpressed AbpA and AbpB or in the absence of AbpA and AbpB to those under normal conditions. MH1 (wild type) or TY0836 (ΔabpA-abpB) cells, or MH1 cells harboring pBR322 or pST4-1, were inoculated into LB medium, and the OD660 was determined every 20 min (Supplementary Fig. S3). In all four combinations, the doubling times were virtually the same, indicating that endogenous or exogenous AbpA and AbpB had no effect on bacterial growth. This result indicates that AbpA and AbpB do not affect bacterial DNA synthesis.
In this study, we first demonstrated that AbpA and AbpB expressed from a plasmid inhibited the propagation of T4, T2, T7 and λ phages (Fig. 1 and Table 1), and that both genes were necessary for this inhibition (Table 2). Consistent with this, chromosomal deletion of either abpA or abpB resulted in an increase of T4 phage progeny production of about 20% (Table 3). Seven ts mutations of T4 gene 41, which encodes a replicative DNA helicase, partially suppressed the growth inhibition by AbpA and AbpB (Fig. 4). AbpA and AbpB inhibited phage DNA replication (Fig. 5), while overexpression and deletion of both genes had no effect on the growth of E. coli cells (Supplementary Fig. S3). From these results, we conclude that AbpA and AbpB function together in anti-phage defense by blocking phage DNA replication. Our observation of an interaction between AbpA and AbpB in vivo (Fig. 2) may point the way to discovering the molecular basis of the function of these proteins.
When AbpA and AbpB were expressed together, DNA synthesis and late gene expression of T4 phage hardly occurred (Figs. 3a and 5). Also, transcription of representative late genes 23 and 24 was markedly reduced (Fig. 3b). In the case of T4 phage, DNA replication is essential for transcription of late genes, because the T4 gene product of 45 (Gp45), the sliding clamp of the T4 DNA replisome, facilitates the recruitment of RNA polymerase to late promoters by interacting with Gp33, the co-activator of T4 late transcription, and Gp55, the T4 sigma factor for late promoter recognition (Herendeen et al., 1989; Geiduschek and Kassavetis, 2010). Therefore, the lack of expression of T4 late genes in the presence of AbpA and AbpB is a secondary effect of a deficiency in DNA replication, and is caused by the inability to form the transcription-ready open complexes of T4 late promoters and the low amount of phage DNA as a template for transcription. AbpA and AbpB are therefore likely to directly target phage DNA replication.
We isolated 11 suppressor mutants of T4 that could grow weakly on cells expressing AbpA and AbpB, and all seven of the ts mutations among these 11 were located within gene 41 (Fig. 4). The middle domain (residues 180–380) of Gp41 (the product of gene 41) (475 aa) functions as a DNA helicase domain, and the suppressor mutations localized to codon 192 or 231 within this domain. It will be important to confirm that the ability to synthesize phage DNA in the presence of AbpA and AbpB is restored, at least partially, by these ts mutations. It will also be important to characterize the remaining four suppressor mutants to elucidate the mechanism of inhibition by AbpA and AbpB. Since we confirmed that there were no mutations in gene 41 of the four non-ts mutants, these suppressors may have arisen from mutations in other gene(s) involved in DNA replication.
abpA and abpB are located in the region of prophage CP4-57, which is a phage genome integrated into the K-12 DNA chromosome (Wang et al., 2009). Genes in prophage regions are generally induced by environmental stress, including phage infection, and may contribute to survival under environmental stress (Wang et al., 2010). For example, nonA of Bacillus subtilis is situated in the region of prophage SPβ, whose genome is 135 kb in length and contains 187 ORFs. This gene is induced by infection with Bacillus phage SP10, and severely inhibits phage DNA replication, resulting in the suppression of phage propagation (Yee et al., 2011). We will therefore monitor the expression level of AbpA and AbpB under variable environmental stress and phage infection. This analysis may explain why the inhibitory effect of both proteins expressed from the K-12 genome was weak (Table 3), whereas their expression from a plasmid completely inhibited phage propagation (Table 1).
AbpA and AbpB inhibited the DNA replication of T4, T7 and λ phages, which belong to three families (Fig. 5), even though the mechanism of DNA replication and replicative factors are quite different among phages. In addition, although λ and E. coli share several replicative factors, DNA replication of E. coli itself escapes inhibition by AbpA and AbpB, as judged from the absence of any apparent effect of AbpA and AbpB on bacterial growth (Supplementary Fig. S3). To address this mystery, we are currently searching for species of phages that can grow on cells expressing AbpA and AbpB. Because some phages in the T-even family, such as RB32 and RB69, can grow in the presence of AbpA and AbpB, a comparison of these phages with T4 may provide clues to solve this mystery. Experiments to examine the interaction between Gp41 and AbpA and AbpB, or the importance of the DEAD-box helicase domain of AbpB in inhibiting phage DNA replication, may also contribute to our understanding of the inhibitory mechanism used by AbpA and AbpB.
In addition to nonA of B. subtilis described above, six lactococcal proteins (AbiA, AbiF, AbiK, AbiP, AbiR and AbiT) have been reported to interfere with phage DNA replication (Chopin et al., 2005), although the details of their functions are still unclear. The present study also indicates that AbpA and AbpB in E. coli are anti-phage factors targeting phage DNA replication. Inhibition of phage DNA replication as an anti-phage mechanism may well be widely conserved among bacteria. Therefore, further study of the inhibitory mechanism of AbpA and AbpB should provide general insights into anti-phage mechanisms that target DNA replication.
We cordially thank Dr. John W. Drake at the U. S. National Institute of Environmental Health Sciences for invaluable help with the manuscript, and the staff of the Radioisotope Research Center at Toyonaka, Osaka University, where all of our experiments using radioisotopes were carried out, for facilitating our research. This work was supported in part by a grant from the program Grant-in-Aid for Young Scientists (B) to Y. O., and in part by a grant from the program Grants-in-Aid for Scientific Research (C) to T. Y., from the Ministry of Education, Culture, Sports, Science and Technology of Japan.