Short communications
A possible packaging signal in the rotavirus genome
2014 Volume 89 Issue 2 Pages 81-86
Details
2014 Volume 89 Issue 2 Pages 81-86
Group A rotavirus (RVA), an etiological agent of gastroenteritis in young mammals and birds, possesses a genome of 11 double-stranded RNA segments. Although it is believed that the RVA virion contains one copy of each genomic segment and that the positive-strand RNA (+RNA) is incorporated into the core shell, the packaging mechanisms of RVA are not well understood. Here, packaging signals of RVA were searched for by analyzing genomic sequences of mammalian and avian RVA, which are considered to have evolved independently without reassortment. Assuming that packaging is mediated by direct interaction between +RNA segments via base-pairing, co-evolving complementary nucleotide sites were identified within and between genomic segments. There were two pairs of co-evolving complementary sites within the segment encoding VP7 (the VP7 segment) and one pair between the NSP2 and NSP3 segments. In the VP7 segment, the co-evolving complementary sites appeared to form stem structures in both mammalian and avian RVA, supporting their functionality. In contrast, co-evolving complementary sites between the NSP2 and NSP3 segments tended to be free from base-pairings and constituted loop structures, at least in avian RVA, suggesting that they are involved in a specific interaction between these segments as a packaging signal.
Rotavirus (RV), a genus in the subfamily Sedoreovirinae of the family Reoviridae (Attoui et al., 2012), is an etiological agent of acute gastroenteritis in young mammals and birds (Bishop et al., 1973). The RV genome consists of 11 segments of double-stranded RNA (dsRNA), all but one of which are monocistronic and encode VP1, VP2, VP3, VP4, VP6, VP7, NSP1, NSP2, NSP3, and NSP4; the eleventh encodes NSP5 and occasionally NSP6 (Estes and Kapikian, 2007). The RV virion is a non-enveloped triple-layered particle; the outer capsid, the inner capsid, and the core shell are composed of VP7 and VP4, VP6, and VP2, respectively. According to the antigenic and genetic properties of VP6, RV is classified into groups A-H (RVA-RVH), among which RVA is the most prevalent (Matthijnssens et al., 2012). RVA is further classified into 27 G (G1–G27) and 37 P (P[1]–P[37]) types based on the properties of VP7 and VP4, respectively, which contain neutralization epitopes (Matthijnssens et al., 2011). Because of a low particle-to-plaque forming unit ratio and an equimolar synthesis of dsRNA and negative-strand RNA (–RNA) in infected cells, the RVA virion is believed to contain one copy of each genomic segment (Hundley et al., 1985; Patton, 1990; Joklik and Roner, 1995; McDonald and Patton, 2011). However, the mechanisms that ensure the selective packaging of RVA are not well understood.
In designing a strategy to elucidate the packaging mechanisms of RVA, it may be helpful to refer to other viruses. The genome of human immunodeficiency virus type 1 (HIV-1) is a non-segmented positive-strand RNA (+RNA), but the HIV-1 virion is known to incorporate two genomic copies that are dimerized (Paillart et al., 2004). Dimerization occurs via base-pairing of a self-complementary stem-loop structure called the dimerization initiation site, where the base-pairing starts at the loop (formation of a kissing loop complex) and extends into the stem (formation of an extended duplex). Influenza A virus (IAV) possesses an eight-segmented –RNA genome, and the IAV virion contains one copy of each genomic segment (Noda et al., 2006; Chou et al., 2012; Fournier et al., 2012). In each segment, the 5’- and 3’-terminal ~20 nucleotides form a panhandle structure via long-range interaction (LRI) (Suzuki and Kobayashi, 2013), and packaging signals are located in the terminal ~200 nucleotides including the untranslated regions (UTRs) and parts of the open reading frames (ORFs), which are segment-specific but highly conserved (Hutchinson et al., 2010). Direct interaction among genomic segments was observed in the IAV virion (Noda et al., 2012). In addition, certain pairs of genomic RNA segments were found to hybridize in vitro, and stem-loop structures that might form a kissing loop complex and an extended duplex were identified in one pair of segments (Gavazzi et al., 2013). Thus, direct interaction between single-stranded RNAs via base-pairing mediated by secondary structures is involved in the selective packaging of HIV-1 and IAV.
For RVA, +RNA bearing a 5’ cap and lacking a 3’ poly(A) tail is known to be incorporated into the core shell (McDonald and Patton, 2011). The +RNA of each genomic segment was inferred to form secondary structures, including a panhandle structure via 5’- and 3’-terminal LRI (Chen and Patton, 1998; Tortorici et al., 2006; Li et al., 2010). Packaging signals that are segment-specific and highly conserved, including UTRs and parts of ORFs, are thought to be located at the 5’ and 3’ termini of genomic segments (Li et al., 2010; McDonald and Patton, 2011). Since many of these characteristics are shared by IAV, the packaging mechanisms of RVA may resemble those of IAV. Indeed, partial complementarity was observed between the 5’- and 3’-terminal ~50 nucleotides of the NSP2 and NSP3 segments, respectively, although an involvement of secondary structures was unclear (Li et al., 2010). Thus, packaging mechanisms of RVA may be studied by identifying complementary nucleotides between genomic segments. The functional relevance of such complementarity may be explored by analyzing conservation of nucleotide sequences and involvement of secondary structures. Co-evolution of complementary nucleotides may also indicate that functional constraint operates on the complementarity (Kimura, 1983, 1985).
If complementarity is involved in the packaging mechanisms of RVA, co-evolution of complementary nucleotides may suppress reassortments between distantly related strains (Heiman et al., 2008; McDonald et al., 2009a, 2009b; McDonald and Patton, 2011; Matthijnssens and Van Ranst, 2012). Reassortment is known to occur within mammalian and avian RVA (Ward et al., 1990; Schumann et al., 2009). However, although interspecies transmission of mammalian RVA to birds and of avian RVA to mammals can occur (Brussow et al., 1992a, 1992b; Mori et al., 2001; Wani et al., 2003; Asano et al., 2011), mammalian and avian RVA are believed to have diverged along with their hosts and to have evolved independently without reassortment (Ito et al., 2001; Trojnar et al., 2009). Reassortment may be suppressed not only by the incompatibility of packaging signals but also by incompatibility in the functions of proteins encoded by genomic segments (Hutchinson et al., 2010; McDonald and Patton, 2011). However, the complementary nucleotides that are involved in packaging, if any, may have diverged between mammalian and avian RVA through co-evolution. The purpose of the present study was to search for packaging signals of RVA in an analysis of the genomic sequences of mammalian and avian RVA.
As of July 18, 2013, complete genomic sequences of RVA were available for 88 mammalian and two avian strains, and were retrieved from the International Nucleotide Sequence Database (INSD) (Supplementary Table S1). Multiple alignment of nucleotide sequences was made for each genomic segment of these 90 strains using the computer program MAFFT (Katoh et al., 2002). Lack of reassortment between mammalian and avian RVA was confirmed by conducting a phylogenetic analysis for each genomic segment (Felsenstein, 1985; Saitou and Nei, 1987; Nei and Kumar, 2000; Tamura et al., 2011); mammalian and avian strains always formed distantly related clusters in phylogenetic trees (Supplementary Figs. S1–S11). Pairs of single nucleotide sites that were complementary in all strains were identified within and between genomic segments. To reduce false-positives in inferences of the functional relevance of complementary nucleotides, complementarity was restricted here to the pairs U and A, and C and G, although U and G may also form a wobble base-pair in RNA. For each pair of single complementary sites, co-evolution was considered to have taken place when the nucleotides observed in mammalian strains were all different from those observed in avian strains, and the proportion of co-evolving sites (pco-evolving) was obtained. Pairs of complementary nucleotide sites longer than 1 were also identified within and between genomic segments. For each length of complementary sites, the probability of containing a particular number or more of co-evolving sites under the null hypothesis of random distribution was computed using the binomial distribution with the probability of occurrence of a co-evolving site given by pco-evolving; e.g., for a pair of complementary sites of length 6 containing three co-evolving sites, the associated probability was given by
The length of complementary nucleotide sites identified within and between genomic segments of RVA ranged from 1 to 7 (Table 1). It should be noted that some of the complementary sites counted in Table 1 are overlapping; e.g., a stretch of complementary sites of length n contains (n – i + 1) stretches of complementary sites of length i (1 ≤ i ≤ n). The pco-evolving value was determined to be 0.00991 (= 65,078/6,564,157), and seven pairs of complementary sites of length ranging from 3 to 6 were judged as containing a significantly large number of co-evolving sites (Table 1). The nucleotide sequences at these sites are presented in Table 2, where the positions are numbered according to the prototype human strain Wa (Wyatt et al., 1980; Wentzel et al., 2013). After eliminating overlapping pairs, there remained three unique pairs of complementary sites: two pairs within the VP7 segment (positions 13–15 [5’GAG3’] and 40–42 [5’CTC3’] and positions 54–59 [5’TGGTAT3’] and 65–70 [5’ATACCA3’]) and one pair between the NSP2 (positions 43–48 [5’AGCCAT3’]) and NSP3 (positions 1050–1055 [5’ATGGCT3’]) segments.
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/89_81_tbl_1.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
|
| Length of complementary sitesa | Number of co-evolving sitesb | Genomic segment | Nucleotide positions in Wa | Sequence in mammalian RVA | Sequence in avian RVA | Genomic segment | Nucleotide positions in Wa | Sequence in mammalian RVA | Sequence in avian RVA |
|---|---|---|---|---|---|---|---|---|---|
| 6 | 3 | NSP2 | 43–48 | 5’AGCCAU3’ | 5’CGUUAU3’ | NSP3 | 1050–1055 | 5’AUGGCU3’ | 5’AUAACG3’ |
| 6 | 2 | VP7 | 54–59 | 5’UGGUAU3’ | 5’UAGUAC3’ | VP7 | 65–70 | 5’AUACCA3’ | 5’GUACUA3’ |
| 5 | 3 | NSP2 | 43–47 | 5’AGCCA3’ | 5’CGUUA3’ | NSP3 | 1051–1055 | 5’UGGCU3’ | 5’UAACG3’ |
| 5 | 2 | NSP2 | 44–48 | 5’GCCAU3’ | 5’GUUAU3’ | NSP3 | 1050–1054 | 5’AUGGC3’ | 5’AUAAC3’ |
| 5 | 2 | VP7 | 55–59 | 5’GGUAU3’ | 5’AGUAC3’ | VP7 | 65–69 | 5’AUACC3’ | 5’GUACU3’ |
| 4 | 3 | NSP2 | 43–46 | 5’AGCC3’ | 5’CGUU3’ | NSP3 | 1052–1055 | 5’GGCU3’ | 5’AACG3’ |
| 3 | 3 | VP7 | 13–15 | 5’GAG3’ | 5’AGU3’ | VP7 | 40–42 | 5’CUC3’ | 5’ACU3’ |
The functional relevance of these sites was explored by analyzing secondary structures of +RNA, which were inferred by RNAFOLD (Lorenz et al., 2011) using the human strain Wa and the pigeon strain PO-13 (Minamoto et al., 1988; Ito et al., 2001) as the reference for mammalian and avian RVA, respectively (Supplementary Figs. S12–S17). In RNAFOLD, base-pairing and non-base-pairing probabilities were assigned to each of base-pairing and non-base-pairing nucleotides, respectively. For complementary sites within the same segment to be functionally relevant, they are expected to form base-pairings with each other. In contrast, complementary sites between different segments are expected to be free from base-pairings within each segment, so that they can interact with each other. Nucleotide sites in each genomic segment of Wa and PO-13 were classified according to whether or not they were involved in base-pairings. Sliding window analysis with a given window size of W and a step size of 1 was conducted for all genomic segments to obtain the empirical probability distribution for the number of non-base-pairing sites contained. A stretch of W nucleotide sites was judged as containing a significantly large number of non-base-pairing sites when the probability of containing the observed number or more of non-base-pairing sites in the empirical probability distribution was below the significance level of 0.05.
In the secondary structure of the VP7 segment, two pairs of complementary sites (positions 13–15 and 40–42, and positions 54–59 and 65–70) formed base-pairings with high probability values in both Wa and PO-13 (Fig. 1, A and B, see also Table 2). Base-pairings of positions 13–15 and 40–42 were also seen in the secondary structure for mammalian RVA reported by Li et al. (2010). In that study, positions 54–59 and 65–70 were both involved in partial base-pairings with 3’-terminal sites via LRI. However, the probability values assigned to these positions were higher in the present study than in Li et al. (2010). Furthermore, both of positions 13–15 and 40–42, and positions 54–59 and 65–70 were also observed to form base-pairings in all other 87 mammalian and one avian strains analyzed in the present study (Supplementary Table S1). Thus, positions 13–15 and 40–42, and positions 54–59 and 65–70 are likely to form stem structures in the 5’ UTR and in the ORF of the VP7 segment, respectively, which appear to be under functional constraint.
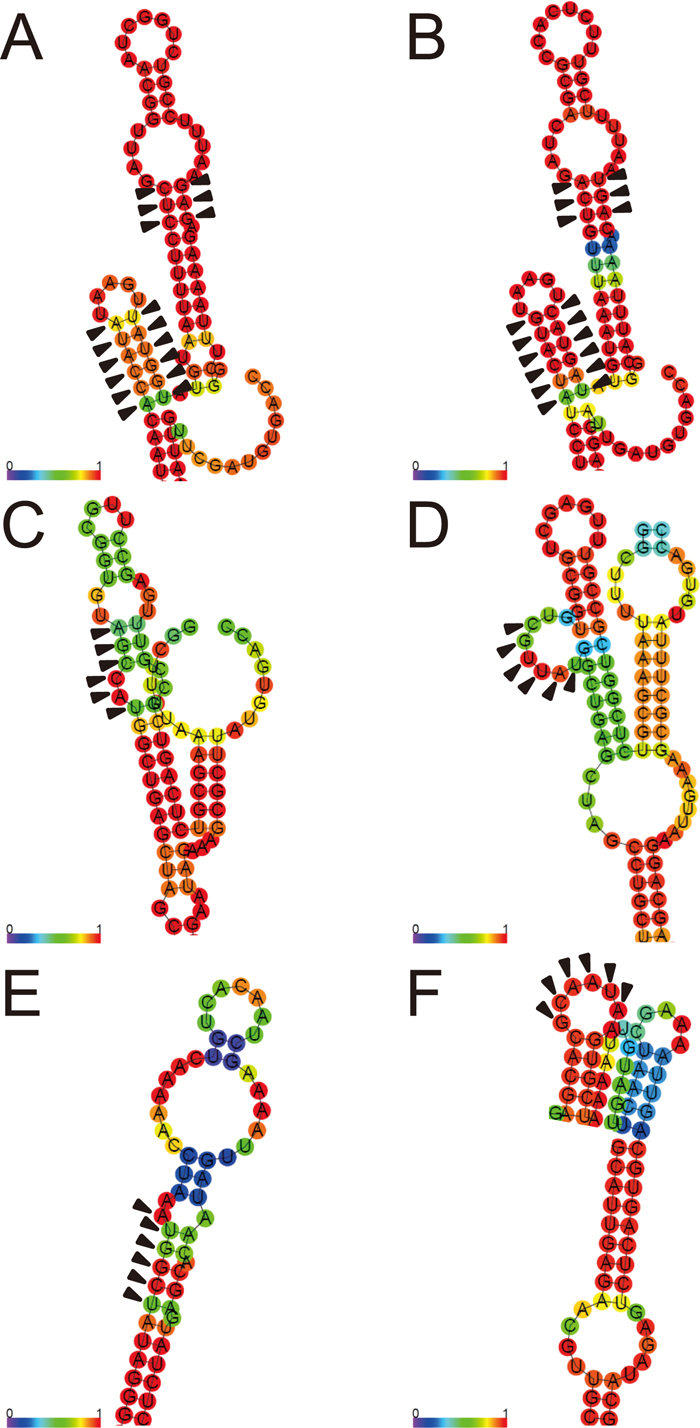
Local secondary structures of +RNA for VP7 segment of (A) Wa (Supplementary Fig. S12) and (B) PO-13 (Supplementary Fig. S13), NSP2 segment of (C) Wa (Supplementary Fig. S14) and (D) PO-13 (Supplementary Fig. S15), and NSP3 segment of (E) Wa (Supplementary Fig. S16) and (F) PO-13 (Supplementary Fig. S17), containing co-evolving complementary nucleotide sites (arrowheads). Secondary structures were inferred using RNAFOLD (Lorenz et al., 2011). Base-pairing and non-base-pairing probabilities are color-coded for base-pairing and non-base-pairing nucleotides, respectively, as indicated by the scale bars.
Regarding positions 43–48 of the NSP2 segment and positions 1050–1055 of the NSP3 segment in Wa, the number of sites that were free from base-pairings was two and one, respectively (Fig. 1, C and E, see also Table 2). The same local structures are also seen in Li et al. (2010). The empirical probability that a stretch of six nucleotides in the Wa genome contains two or more, or one or more non-base-pairing sites was 0.616 and 0.833, respectively. The joint probability that, in a pair of stretches of six nucleotides, one contains two or more, and the other contains one or more non-base-pairing sites can be obtained by subtracting from unity the probabilities that either stretch does not contain a non-base-pairing site and that both stretches contain one non-base-pairing site; this yielded a joint probability value of 0.647. Therefore, these sites did not tend to be free from base-pairings. It should be noted, however, that the probability values assigned to the base-pairing positions were relatively low in both NSP2 and NSP3 segments. In contrast, in PO-13, all of six complementary sites were free from base-pairings and constituted loop structures in both NSP2 and NSP3 segments, with high probability values (Fig. 1, D and F, see also Table 2). The empirical probability that all sites in a stretch of six nucleotides in the PO-13 genome were free from base-pairings was 0.0997, and the joint probability for two stretches was 0.00994 (= 0.09972). Thus, these sites tended to be free from base-pairings, at least in avian RVA.
When the secondary structures of +RNA for the NSP2 and NSP3 segments were examined in all other 87 mammalian and one avian strains, both of positions 43–48 of the NSP2 segment and positions 1050–1055 of the NSP3 segment were free from base-pairings in the avian strain (Supplementary Table S1), confirming the above results. However, it was also observed that positions 43–48 of the NSP2 segment were free from base-pairings in four human strains and that positions 1050–1055 of the NSP3 segment were free from base-pairings in three human, three horse, three cow, and one antelope strains (Supplementary Table S1). These strains were sparsely distributed in phylogenetic trees (Supplementary Figs. S1–S11), suggesting that the co-evolving complementary nucleotide sites in the NSP2 and NSP3 segments also form loop structures in mammalian RVA under certain conditions.
Positions 43–48 of the NSP2 segment and positions 1050–1055 of the NSP3 segment both reside in the terminal ~200 nucleotides, where packaging signals are thought to be located (Li et al., 2010; McDonald and Patton, 2011). Furthermore, these positions are included in the 5’- and 3’-terminal ~50 nucleotides of the NSP2 and NSP3 segments, respectively, which were reported to be partially complementary (Li et al., 2010). In that study, however, these positions were inferred to form partial base-pairings with different positions of the other segment; moreover, the involvement of secondary structures in the formation of a possible duplex of ~50 nucleotides was unclear. Considering the packaging mechanisms of HIV-1 and IAV, and the similarities in the characteristics related to packaging between IAV and RVA, it is possible that the loop structures containing positions 43–48 of the NSP2 segment and positions 1050–1055 of the NSP3 segment form a kissing loop complex and facilitate the formation of an extended duplex of ~50 nucleotides. It would be interesting to conduct experiments to test these hypotheses. Although the last two sites of positions 43–48 of the NSP2 segment constitute the first two sites of the start codon in both Wa and PO-13, the remaining sites exist in the 5’ UTR. In addition, all of positions 1050–1055 of the NSP3 segment are located in the 3’ UTR in both strains. It is therefore of interest to examine whether packaging is impaired by disrupting the complementarity, and subsequently rescued by recovering the complementarity, in different pairs of nucleotides through site-directed mutagenesis without affecting the functions of the encoded proteins (Komoto et al., 2006).
In the present study, a possible packaging signal of RVA was identified that may be involved in the specific interaction between the NSP2 and NSP3 segments. Theoretically, however, at least 10 packaging signals are required for selective packaging of 11 genomic segments, and therefore many of them remain to be discovered. This may be due to limitations in the current strategy, where several assumptions about packaging signals were made to facilitate detection from statistical analyses of genomic sequences. First, selective packaging was assumed to be mediated by direct RNA-RNA interaction via base-pairing of U and A, and C and G without gaps. Second, nucleotide positions involved in packaging signals were assumed to have been maintained during the evolution of mammalian and avian RVA. Third, co-evolution of complementary nucleotide sites was assumed to have occurred on the lineages separating mammalian and avian RVA. Fourth, nucleotide positions involved in packaging signals were assumed to be free from base-pairings and to form a loop structure. These strict assumptions may have decreased the sensitivity of detecting packaging signals, although they may have increased the specificity. Some of these assumptions may also be violated; for example, it has been proposed that nucleotide positions involved in packaging signals in IAV change during evolution (Gavazzi et al., 2013). In addition, even if these assumptions were satisfied, packaging signals should be long and have accumulated a number of compensatory nucleotide substitutions to be detectable by statistical analyses. It may be important to improve the strategy (e.g., by incorporating the pattern of nucleotide substitution) and to relax the assumptions (e.g., by allowing for wobble base-pairs, mismatches, and gaps) to detect the missing packaging signals of RVA.
The author thanks two anonymous reviewers for valuable comments. This work was supported by a Grant-in-Aid for Research in Nagoya City University to Y. S.