Special reviews
Emerging links between iron-sulfur clusters and 5-methylcytosine base excision repair in plants
2016 Volume 91 Issue 2 Pages 51-62

Details
2016 Volume 91 Issue 2 Pages 51-62
Iron-sulfur (Fe-S) clusters are ancient cofactors present in all kingdoms of life. Both the Fe-S cluster assembly machineries and target apoproteins are distributed across different subcellular compartments. The essential function of Fe-S clusters in nuclear enzymes is particularly difficult to study. The base excision repair (BER) pathway guards the integrity of DNA; enzymes from the DEMETER family of DNA glycosylases in plants are Fe-S cluster-dependent and extend the BER repertowere to excision of 5-methylcytosine (5mC). Recent studies in plants genetically link the majority of proteins from the cytosolic Fe-S cluster biogenesis (CIA) pathway with 5mC BER and DNA repair. This link can now be further explored. First, it opens new possibilities for understanding how Fe-S clusters participate in 5mC BER and related processes. I describe DNA-mediated charge transfer, an Fe-S cluster-based mechanism for locating base lesions with high efficiency, which is used by bacterial DNA glycosylases encoding Fe-S cluster binding domains that are also conserved in the DEMETER family. Second, because detailed analysis of the mutant phenotype of CIA proteins relating to 5mC BER revealed that they formed two groups, we may also gain new insights into both the composition of the Fe-S assembly pathway and the biological contexts of Fe-S proteins.
Fe-S clusters are prosthetic groups composed of iron and sulfur, ligated mainly to cysteine residues in their associated proteins. They are involved in a myriad of processes across all kingdoms of life where they participate in important biological functions, among which respiration and photosynthesis were discovered first. The most common types of Fe-S clusters are rhombic [2Fe-2S], e.g., in ferredoxin in chloroplasts and mitochondria, and cubane [4Fe-4S], e.g., in nuclear enzymes (Beinert et al., 1997; Netz et al., 2014). Fe-S clusters are very versatile; their functions can be broadly grouped into electron carriers, enzymatic catalysts and gene regulators.
Fe-S clusters are associated with the origin of life in the ‘iron-sulfur world’ theory (Wächtershäuser, 2007), consistent with the wide distribution of Fe-S proteins in all domains of life. Experimental detection of Fe-S proteins is still cumbersome, despite developments in spectroscopic techniques (Rouault, 2015), and universal consensus sequences for ligation sites of Fe-S clusters could not be found. It is therefore expected that many Fe-S proteins are currently unrecognized (Fontecave, 2006; Rouault, 2015). RNA polymerase is an example of an enzyme that was found to possess an Fe-S cluster (Hirata et al., 2008) long after its first discovery. Low estimates indicate that 3% of the proteins in Escherichia coli are Fe-S cluster-dependent (Py and Barras, 2010) and 100 Fe-S proteins exist in plants (Balk and Pilon, 2011).
An important class of enzymes recently recognized as Fe-S proteins have functions in DNA metabolism: DNA glycosylases were identified as such some time ago (Kuo et al., 1992), while helicases, nucleases, primases and polymerases have subsequently also emerged (White and Dillingham, 2012). Investigation of the precise molecular function of the Fe-S cluster in these essential proteins in vivo is particularly challenging and has remained elusive.
Fe-S assembly machineriesDespite the simple composition of Fe-S clusters, their assembly and delivery into apoproteins is elaborated by a number of multi-protein systems. In bacteria, there are three Fe-S assembly pathways: ISC (iron-sulfur cluster) is considered the housekeeping system and SUF (sulfur mobilization) the stress-dedicated system (Py et al., 2011), while NIF (nitrogen fixation) is required for maturation of nitrogenase in nitrogen-fixing bacteria. Eukaryotic cells host Fe-S assembly pathways of bacterial origin in mitochondria (ISC-like) and in chloroplasts (SUF-like) and, therefore, these pathways are highly conserved across the bacterial, plant and animal kingdoms (Schilke et al., 1999; Balk and Lobreaux, 2005; Ye et al., 2006; Lill and Muhlenhoff, 2008). In addition, an assembly pathway specific to eukaryotes exists, namely CIA (cytosolic iron–sulfur protein assembly) (Lill and Muhlenhoff, 2008; Lill, 2009; Xu and Moller, 2011). Since its discovery in the early 2000s, intense investigations by R. Lill’s group have led to the identification of many components, especially in yeast but also in the human CIA pathway (Paul and Lill, 2015). The CIA pathway is responsible for the maturation of cytosolic and nuclear Fe-S proteins, and deficiency in CIA components interferes with cell growth in yeast and viability in plants and mammals (Couturier et al., 2013; Netz et al., 2014). Remarkably, the CIA pathway is dependent on the mitochondrial ISC-like pathway (see below), thus making the function of both cytosolic and nuclear Fe-S proteins dependent on mitochondria.
Like all other Fe-S assembly pathways, CIA includes scaffold and carrier proteins. In this paragraph, the yeast nomenclature is used unless otherwise specified. First, iron, sulfur and electrons are needed for de novo assembly onto scaffold proteins. Whereas the origin of iron is not clear, sulfur is immobilized by a cysteine desulfurase in the mitochondria. The CIA pathway obtains a sulfur precursor from ISC via the export component ABC transporter Atm1p and the sulfhydryl oxidase Erv1p (Kispal et al., 1999; Lange et al., 2001). The flavoprotein Tah18p and Dre2p protein function as a donor complex for NADPH-derived electrons in an early CIA step (Netz et al., 2010). Next, the nucleotide binding protein Nbp35p and Cfd1p cooperate as scaffold proteins to assemble the Fe-S cluster (Roy et al., 2003; Hausmann et al., 2005). Green algae and plant genomes, however, lack Cfd1 orthologs, and instead, NBP35, the Nbp35p plant ortholog, functions as a homomultimer (Bych et al., 2008). The Fe-S cluster binds in a labile manner to the scaffold proteins and is inserted stably into carrier proteins at a late CIA step. The hydrogenase-like protein Nar1p, WD40 protein Cia1p, DUF59 family protein Cia2p and HEAT repeat domain-containing protein Mms19p form a carrier complex, which is also responsible for the incorporation of the cluster into target apoproteins (Balk et al., 2004, 2005; Srinivasan et al., 2007; Gari et al., 2012; Stehling et al., 2012). Notably, several CIA components, including Cdf1p, Nbp35p, Dre2p and Nar1p, also themselves have Fe-S cluster(s) (Balk et al., 2005; Hausmann et al., 2005; Netz et al., 2010).
Although the roles of each CIA component have not yet been fully elucidated, information about interactions between CIA members at different steps of the pathway is emerging. For example, the conserved Dre2p-Tah18p complex is known to assemble not only Fe-S clusters but also another type of cofactor, namely the di-iron cluster, required by ribonucleotide reductase, which supplies the building blocks for DNA replication and repair (Zhang et al., 2014). Another example is the so-called CIA targeting complex composed of late-acting CIA proteins. According to studies in human, CIA1, CIA2 and MMS19 orthologs (CIAO1, CIA2A, CIA2B/MIP18 and MMS19) potentially form a protein complex (Ito et al., 2010; Netz et al., 2012; van Wietmarschen et al., 2012) and, moreover, the human Nar1 ortholog IOP1 interacts weakly with this complex (Ito et al., 2010; Seki et al., 2013). Interestingly, the components required for Fe-S cluster incorporation differ depending on the target apoprotein (Stehling et al., 2012; Stehling and Lill, 2013).
Alternative maturation pathways for the Fe-S protein Dre2Given that many Fe-S proteins are likely still unrecognized, the Fe-S assembly pathways are likely incompletely elucidated. The conserved eukaryotic protein Dre2 is an antiapoptotic factor in mammalian cells (Shibayama et al., 2004). It can shuttle from cytosol to nucleus upon stimulation with reactive oxygen species in dopaminergic neurons (Park et al., 2011), and can be trapped in the mitochondrial intermembrane space in yeast (Banci et al., 2011). It was intensively studied as an early CIA component in the cytosol and was until very recently an ‘orphan’ Fe-S protein. The assembly of Fe-S clusters onto Dre2 was reported to be independent of CIA components, a mechanism referred to as “unprecedented for a non-scaffold Fe-S protein” (Netz et al., 2010). Studies by Banci et al. have recently elucidated not one, but two alternative maturation pathways for Dre2 in the cytosol (Banci et al., 2015a, 2015b).
Monothiol glutaredoxins (GRXs) can assist Fe-S maturation in both cytosol and mitochondria by acting as 2Fe-2S cluster donors. GRX3 is an in vivo Dre2 binding partner in both yeast (Rual et al., 2005) and mouse (Saito et al., 2011) in the cytosol. A specific interaction between the N-terminal domains of the two proteins promotes the transfer of two 2Fe-2S clusters from GRX3 (Banci et al., 2015b) or GRX3-BOLA2 (Banci et al., 2015a) to Dre2. It was suggested that these two pathways may be operative under normal and oxidative stress conditions, respectively (Banci et al., 2015a).
The CIA pathway serves a broad range of Fe-S proteins, most of which may have a single destination, and perhaps function, in either the cytosol or the nucleus. The Fe-S Dre2 protein appears to fall into a different category: its maturation involves proteins belonging to the iron trafficking/ISC apparatus, rather than the cytosolic Fe-S biogenesis apparatus, and it may have different functions according to its localization to mitochondria, cytosol and nucleus.
In most eukaryotes, methylation can confer additional functions on DNA, including long-term silencing as well as genome-wide reprogramming and reversible responses of specific loci to their stimuli. The latter functions can be supported by enzymatic removal of methylation (Wu and Zhang, 2010). Such active DNA demethylation is one of the DNA-related processes emerging as Fe-S cluster- or iron-dependent, via diverse enzymes as described below. This section is concerned particularly with the direct excision of 5-methylcytosine (5mC) by the base excision repair (BER) pathway in plants, because this activity represents an extension of the established repertowere of this repair pathway. I provide a brief overview of BER before focusing on 5mC BER by Fe-S cluster-dependent DNA glycosylases and the developmental contexts under study in plants.
Fe-S cluster/iron requirements in active DNA demethylationMultiple pathways for active DNA demethylation have been reported in different organisms. The methylated base rather than the methyl group may be directly removed by DNA glycosylases via the BER pathway (an overview is presented below). The helix-hairpin-helix (HhH) DNA glycosylase structural family is widespread in all life kingdoms (Denver et al., 2003; Dalhus et al., 2009). At least two types of HhH glycosylases, both containing four cysteines capable of ligating Fe-S clusters, engage in DNA demethylation via BER: the N-methylpurine glycosylase II that removes 3-methyladenine and 7-methylguanine in a hyperthermophilic bacterium (Begley et al., 1999), and the plant DNA glycosylase family represented by DEMETER that directly excise 5mC to initiate BER (Choi et al., 2002).
Another active DNA demethylation pathway is proposed to involve the elongator protein 3 (ELP3 in mouse) (Okada et al., 2010; Wu and Zhang, 2010), initially identified as a core enzyme of the elongating form of RNA polymerase II complex (Otero et al., 1999). ELP3 belongs to the radical S-adenosylmethionine (SAM) family enzymes, in which a 4Fe-4S cluster is ligated by three cysteines, often from a Cx3Cx2C motif. Fe-S radical SAM enzymes perform reductive cleavage of SAM into 5’-deoxyadenosyl 5’-radicals used in different reactions, and transfer electrons to 4Fe-4S clusters. Currently there is no direct biochemical evidence for how these activities may contribute to the enzymatic action of ELP3 in DNA demethylation. The presence of the 4Fe-4S cluster and possible cleavage of SAM was demonstrated for a bacterial Elp3 (Paraskevopoulou et al., 2006). The Fe-S SAM domain of ELP3 in mouse is required for global DNA demethylation in the paternal genome shortly after the sperm fertilizes the egg (Okada et al., 2010). Also, in Arabidopsis, ELP2, another subunit of the same complex, was implicated in modulating DNA methylation at some genes during the immune response and also across the genome (Wang et al., 2013).
Evidence for direct 5mC BER pathways outside plants is not compelling (Wu and Zhang, 2010). Instead, BER can restore unmethylated cytosines following either i) deamination of methylcytosines to thymines, creating T:G mismatches that are removed by thymine DNA glycosylase (TDG), from another structural glycosylase family (Cortazar et al., 2011; Cortellino et al., 2011); or ii) oxidation of methylcytosine to 5-hydroxymethylcytosine, then to 5-formylcytosine, and finally to 5-carboxylcytosine by the mammalian TET (ten-eleven translocation methylcytosine dioxygenase) family dioxygenases (He et al., 2011). While Fe-S clusters do not seem to be involved in these two pathways, iron is required for oxidative demethylation by TET. Also, the dioxygenase AlkB from E. coli is another example of iron-dependent oxidative demethylation, in this case of 1-methyladenine to 3-methylcytosine.
Brief overview of base excision repairThe BER pathway guards DNA integrity by eliminating single-base lesions-that result from alkylation, oxidation and deamination reactions in the cellular environment. BER is a multistep process. It is initiated by a DNA glycosylase, specialized for a small set of lesions, which excises the damaged base, and it then proceeds via the concerted action of additional proteins, finally restoring the unmodified state.
Recognition. Little is known about how DNA glycosylases can detect base lesions with high accuracy; this task is extraordinarily challenging because the differences between modified and natural bases are very subtle and the glycosylases are often present at low levels. A three-dimensional search termed ‘facilitated diffusion’ is associated with DNA glycosylase recognition (Zharkov and Grollman, 2005), and chromatin based-mechanisms are also believed to impact this step (Odell et al., 2013; Rodriguez et al., 2015). For more sensitive detection of irregularities, selected bases are flipped out of the helix into a selective pocket for substrate interrogation in the subsequent step.
Excision and repair synthesis. DNA glycosylase generates a cleavage in the N-glycosidic bond between the base and the sugar-phosphate backbone of the DNA, generating an abasic site (AP). This can be further addressed in two ways: short- or long-patch BER. In short-patch BER, monofunctional glycosylases require the intervention of an AP endonuclease to generate compatible ends for DNA polymerase β, and then of a DNA ligase to replace a single nucleotide (Jacobs and Schar, 2012). In long-patch BER, bifunctional glycosylases couple base excision with an AP-lyase step, and DNA polymerase δ/ε carry out repair to introduce 2–6 nucleotides, also sealed by a DNA ligase (Jacobs and Schar, 2012). The processes of BER excision and repair synthesis are conserved from bacteria to mammals.
DNA-mediated charge transfer. A popular hypothesis on how Fe-S DNA glycosylases can increase the efficiency of damage detection based on the redox status of their Fe-S cluster is elaborated by work from J. K. Barton’s group (Boal et al., 2007; Sontz et al., 2012; Grodick et al., 2014). In brief, a unique property of well-stacked base pairs of the DNA duplex is to allow charge transfer (CT) over long distances. Redox-active 4Fe-4S cluster BER enzymes are proposed to use this chemistry to cooperatively probe the integrity of the DNA during the initial search for damage in the genome (Grodick et al., 2014; Arnold et al., 2016). The DNA-binding affinity of a protein that has a 4Fe-4S cluster depends on the oxidation state of the cluster: in the oxidized state, the enzyme binds tightly to DNA; in the reduced state, the enzyme dissociates from DNA. DNA-mediated CT requires oxidation of the 4Fe-4S cluster of a protein initially bound to DNA in a non-specific manner. When the bases are normally stacked, CT is facilitated and electron exchange with another 4Fe-4S cluster protein occurs, resulting in the reduced state of the second protein, which causes its dissociation from the DNA. Repair proteins do not accumulate in surrounding regions where there is no lesion. In contrast, when a lesion perturbs the DNA duplex, it represents a CT barrier as well as leading to oxidation of the 4Fe-4S cluster to keep the repair proteins tightly bound to DNA and clustered in its proximity. Enzymes already implicated in this process include two bacterial BER glycosylases, endonuclease III (EndoIII, which targets oxidized pyrimidines) and MutY (which repairs oxoguanine-adenine mismatches), as well as the nucleotide excision repair helicase XPD (which unwinds damaged DNA for repair) and the ATP-dependent helicase DinG (which repairs R-loops) (Grodick et al., 2014).
5mC base excision repair by DEMETERsFour DNA glycosylases in Arabidopsis, DEMETER, REPRESSOR OF SILENCING1 (ROS1), DEMETER-LIKE 2 (DML2) and DEMETER-LIKE 3 (DML3) (called DEMETERs hereafter), form a small gene family containing the conserved HhH motif and an iron-sulfur cluster loop (FCL) motif responsible for 4Fe-4S cluster binding, both of which are also found in E. coli adenine DNA glycosylase MutY and EndoIII (Kuo et al., 1992; Guan et al., 1998; Mok et al., 2010). Nevertheless, DEMETER proteins are unusually large for this class of proteins, with unique protein domains, and they have a function beyond canonical DNA repair in excising 5mC from CG, CHG and CHH contexts (where H is A, G or T) and replacing it with unmethylated cytosine via BER (Choi et al., 2002; Gehring et al., 2006; Morales-Ruiz et al., 2006). DEMETERs are bifunctional glycosylases (Choi et al., 2002; Gong et al., 2002), and are expected to cooperate with DNA polymerase δ/ε in long-patch BER, especially because plant genomes lack sequences encoding DNA polymerase β homologs (Uchiyama et al., 2008), which are characteristic for short-patch BER. Nevertheless, some homologous plant proteins involved in short-patch BER steps (such as X-ray repair cross-complementing protein 1 (XRCC1), AP endonuclease 1 (APE1), 3’-phosphatase zinc finger DNA 3’-phosphoesterase (ZDP) and DNA ligase 1 (AtLIG1) have been identified as the components affected in DNA demethylation (Andreuzza et al., 2010; Li et al., 2015a; Li et al., 2015b). The identity of all BER enzymes downstream of DEMETERs still needs to be revealed.
Developmental contexts of 5mC BER. In plants, conspicuous 5mC BER coupled with transcriptional activation appears to be confined to tissues or even single cells programmed to terminate development shortly after peaks of DEMETER expression (Fig. 1).
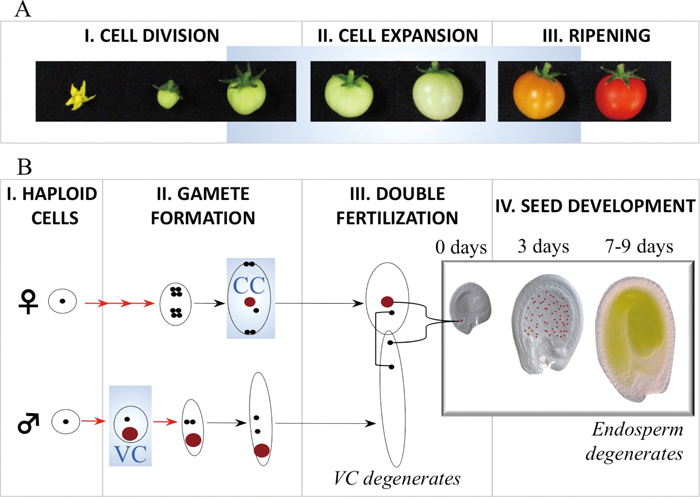
Terminal cellular lineages manifesting conspicuous 5mC BER in plants. A. Tomato fruit develops from the ovary of the flower, and undergoes cell division and cell expansion phases before it ripens (stages I–III) and finally senesces (not shown). Pictures courtesy of Hiroshi Ezura and Mariko Takayama. B. Post-meiotic divisions (red arrows) of female and male haploid cells lead to the formation of differentiated cells including the central cell (CC) of the embryo sac and the paternal vegetative cell (VC) of the pollen (both represented as red circles) with accessory roles in fertilization. Double fertilization is indicated by a square bracket (egg cell fusion with one sperm cell) and a brace bracket (central cell fusion with one sperm cell). On fertilization, the CC divides to give rise to the endosperm, a placenta-equivalent tissue that nourishes the embryo in the first 7–9 days in Arabidopsis (in the white insert panel, CC and endosperm nuclei are marked in red), and the VC functions to extend the pollen tube carrying the sperm cells to the embryo sac. Stages where DNA demethylation takes place are highlighted with a blue background.
In tomato, SlDML2, one of the four DEMETERs, is highly expressed in the fruit pericarp (Liu et al., 2015), a tissue enclosing the seeds whose main function is to delay the maturation of the fruit until the seeds become mature, under the control of the plant hormone ethylene. Two major phases of ethylene production, accompanied by global gene expression changes, are marked by the transition from a phase of cell division to cell expansion when ripening proceeds until fruit eventually senesces (Fig. 1A). SlDML2 was recently reported to demethylate DNA at the onset of ripening in the pericarp (Zhong et al., 2013; Liu et al., 2015). This macroscopic tissue may be very useful for more detailed in vivo biochemical studies of BER 5mC excision in the future.
DEMETER has been most intensively studied in the model plant Arabidopsis in microscopic structures. Upon fertilization, DEMETER is highly expressed in two distinct single cells (Fig. 1B): the central cell (CC) of the embryo sac (Gehring et al., 2006) and the vegetative cell (VC) of the pollen (Schoft et al., 2011), which also expresses ROS1. Fertilization in flowering plants proceeds from two fusions of gametes: the egg cell with one sperm cell, and the CC with another sperm cell. The first fusion forms the next-generation lineage (embryo to mature plant), while the second fusion forms an embryo-nourishing placenta-like tissue, the endosperm. The two sperm cells are transported to the female gametes through an elongating tube generated by the VC of pollen. Both the endosperm and the pollen tube are terminal structures and therefore their progenitor cells, the CC and the VC, may be considered as accessory cells. Notably, genome-wide DNA methylation was analyzed in isolated VCs and their precursors in wild type and dme (Calarco et al., 2012) but, so far, not in isolated CCs. The experimental system from which the CC DNA methylome is inferred is the seven-day-old endosperm (Fig. 2).
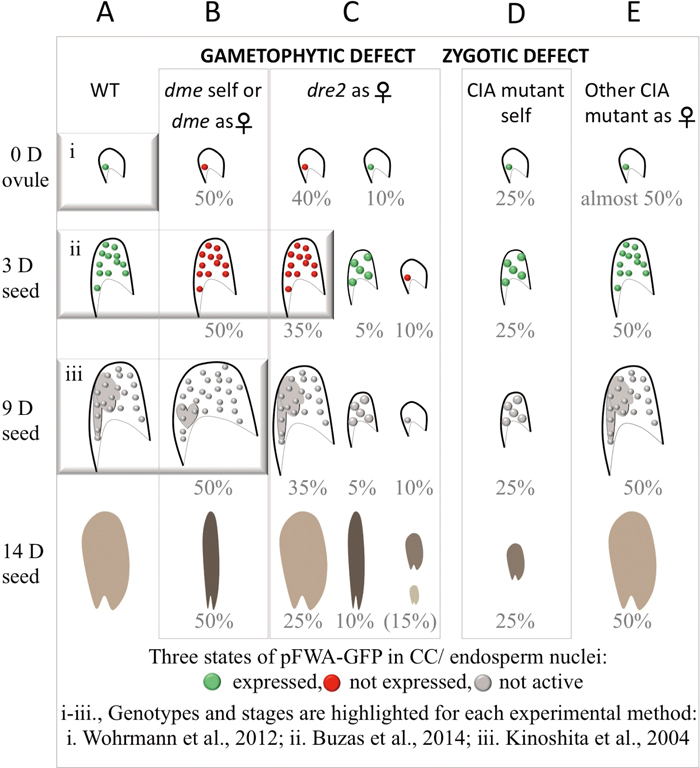
The CC/endosperm experimental system and mutant phenotypes. (Note: mutant nomenclature is designated by lower-case italics.) A. In wild type, pFWA-GFP is expressed (green circles) in the unfertilized central cell (0 days, 0 D) and its progeny, the endosperm, up to three days after fertilization (3 D). The pFWA promoter is inactive after this stage (gray circles at nine days after fertilization (9 D). Coordinated development between the embryo and endosperm can be observed, for example at 9 D, and seeds are fully mature and desiccated at 14 D. B. The maternal gametophytic function of DEMETER can be monitored when the mutant allele is maternal or in self–fertilized siliques (the transmission of the paternal allele is almost null) from 0 D to 3 D in dme using pFWA-GFP. All the ovules and seeds that inherit dme (50%) do not express pFWA-GFP (red circles), in contrast to the segregating wild type ones. Otherwise, no morphologic abnormalities can be observed at these stages. At about 9 D, the dme endosperm is overproliferating, causing the seed to enlarge, and the embryo is underdeveloped. This phenotype allows visual identification of mutant seeds, all of which will collapse by 14 D (i.e., the phenotype is fully penetrant). C. The maternal gametophytic function of AtDRE2 can be detected when the mutant allele is maternal by the lack of pFWA-GFP fluorescence from 0 D to 3 D. A notable difference compared to dme is that the phenotype is not fully penetrant and varies during development (40% inactivation at 0 D and 35% inactivation at 3 D). Additionally, about 15% of the seeds (including unfertilized ovules) have early developmental defects. Later in development, the seeds that were unaffected early in development appear normal, although, upon dissection of 14 D seeds, embryos smaller than wild type can be identified. Only about 10% of seeds at 14 D abort in a similar manner to dme seeds. D. All CIA mutants tested, including atdre2-2 and the above-mentioned alleles, share a similar early abortion zygotic seed defect, which can be detected in selfed plants. E. When the mutant allele was maternal, other CIA mutants tested did not manifest a clear pFWA-GFP or seed development phenotype like dme and atdre2. These included the following alleles: atatr3-3, atnpb35-1, cia1-1 (Buzas et al., 2014) and nar1-3 (Nakamura et al., 2013; Buzas et al., 2014).
The majority of plant body cells are dividing somatic cells that express three DEMETERs. Here, 5mC BER has a limited effect on gene expression (Penterman et al., 2007). When somatic cells are stimulated with pathogen elicitors, modest DNA demethylation changes can be detected in intact plants (Yu et al., 2013; Le et al., 2014). A case was also reported where ROS1 activity was required for differentiation of a specific cellular type in leaf epidermis. To explain how such specificity could be achieved, it was suggested that ROS1 function could be regulated by environmental or developmental cues (Yamamuro et al., 2014).
At least four proteins from the CIA pathway in plants have recently been associated with DNA demethylation- and repair-related processes (Luo et al., 2012; Nakamura et al., 2013; Buzas et al., 2014; Duan et al., 2015; Han et al., 2015). These findings emerged mainly from two types of genetic screens, outlined below, targeted to isolate new factors promoting DNA demethylation at either DEMETER or ROS1 target sites in the genome. I detail the complex action of DEMETER in one maternal gamete and in the maternal endosperm, to help understand the mutant phenotypes of AtDRE2 and AtNAR1 and their putative roles in Arabidopsis. I also summarize the role of proteins from the CIA targeting complex in multiple nuclear processes including 5mC BER and DNA repair in somatic cells.
The central cell/endosperm-based experimental systemDEMETER was named after the goddess of grains and fertility of the earth to reflect its role in seed development. Seeds are complex, highly dynamic and, in Arabidopsis, tiny structures. They enclose two highly coordinated lineages from the time of differentiation of their progenitor cells to either maturation (the embryo) or termination (the endosperm). The maternal dme mutant allele causes seed abortion; this phenotype is due to the early expression of DEMETER in unfertilized CCs but it is manifested as a late developmental imbalance in the seed: the endosperm overproliferates and the embryo remains underdeveloped (Choi et al., 2002).
The overproliferating dme endosperm, distinguishable within dissected siliques without clearing, is a phenotypic hallmark that inspired the development of a manual, microscopic technique for separating the endosperm from the rest of the seed (Fig. 2iii) (Kinoshita et al., 2004). Using this technique, the promoter of a maternally expressed gene named FWA was profiled for DNA methylation: maternal endosperm was hypomethylated relative to paternal endosperm, a feature absent in dme endosperm (Kinoshita et al., 2004). Similar parent-of-origin expression and DNA methylation patterns were later identified in many other genes in manually dissected endosperm from wild type and dme (Gehring et al., 2009; Hsieh et al., 2009). Two assays addressing methylation status before 7-day endosperm were used to date. A quantitative assay based on a methylation-sensitive enzyme in whole seeds at 3 days showed hypermethylation in dme relative to wild type at four genes including FWA (Fig. 2ii). Data from isolated CCs indicating that the FWA promoter may be highly methylated in differentiated CCs (Fig. 2i; Wohrmann et al., 2012) cannot currently be reconciled with the rest of the findings.
DEMETER modus operandi in the CC. The current view on the modus operandi of DEMETER is mainly based on events in two developmental snapshots, separated by seven days from each other: the expression of DEMETER in the mature CC, and endosperm methylation at DEMETER target genes, as follows. DEMETER briefly governs 5mC BER in the CC at the time when it is fully differentiated but before the arrival of the fertilizing sperm. Such brief action is revealed immediately by the activation of maternal gene expression at 5mC excision sites, which in turn may persist for some time in the endosperm lineage due, presumably, to propagation of unmethylated states. The paternal counterpart sites remain hypermethylated and transcriptionally inactive, based on their origin from a sperm cell not exposed to 5mC BER. Thus, the DNA methylation differences found between the maternal and paternal endosperm genomes are explained by the action of DEMETER in the CC.
FWA-GPF-based screening. DEMETER is believed to be the only enzyme that excises 5mC in the central cell, because maternal DNA demethylation is nearly fully reversed in dme mutant endosperm (Gehring et al., 2009; Hsieh et al., 2009). FWA is expressed during the first three days after fertilization in wild type but not in dme (Fig. 2, A and B). Therefore, it is assumed that FWA expression requires 5mC removal by DEMETER in the CC. A reporter fusion with the FWA promoter region (pFWA-GFP) proved to be an invaluable screening tool in identifying other components related to DEMETER DNA demethylation. Because the screen is based on a gametophytic phenotype, even mutants in essential components can be isolated (Ikeda et al., 2011; Nakamura et al., 2013; Buzas et al., 2014). However, mutations in two CIA components could not be analyzed using the manual endosperm separation technique (Kinoshita et al., 2004) because either the mutant phenotype at seven days has low penetrance, in the case of AtDRE2 (Buzas et al., 2014), or it is not manifested in late endosperm, in the case of AtNAR1 (Nakamura et al., 2013).
Role of AtDRE2 and AtNAR1 in gametes and seed developmentMost CIA proteins are required for early embryo development in Arabidopsis (Bych et al., 2008; Luo et al., 2012; Bernard et al., 2013; Nakamura et al., 2013; Buzas et al., 2014) and this function is zygotic (Nakamura et al., 2013; Buzas et al., 2014). In addition, mutations in AtDRE2 and AtNAR1 were identified in the pFWA-GFP screen (Nakamura et al., 2013; Buzas et al., 2014). GFP was not expressed in most CCs inheriting atdre2-2, and was diminished in some atnar1-3 CCs. Similar to dme seeds, the presence of a maternal atdre2 allele causes lack of pFWA-GFP expression and seed abortion, even though these phenotypes are not fully penetrant (Fig. 2, B and C). In contrast, atnar1-3 endosperms pollinated with wild type pollen fully restore pFWA-GFP expression and seed development to wild type. Because the atdre2-2 endosperm does not overproliferate, seeds with morphologically normal atdre2-2 endosperm could only be selected using pFWA-GFP up to three days after fertilization. At this early stage, regions of four DEMETER target genes were hypermethylated in atdre2-2 compared to wild type, indicating that AtDRE2 promotes DNA demethylation (Buzas et al., 2014). Unlike dme and atdre2, GFP fluorescence cannot be used to select atnar1-3 seeds. Therefore, it remained unclear if/how AtNAR1 affects DNA methylation in seeds. Mutants of the CIA proteins ATR3, NPB35 and CIA1, considered non-redundant, affected pFWA-GFP fluorescence in a reduced percentage of CCs and did not have detectable maternal seed defects (Fig. 2, D and E) (Buzas et al., 2014). In addition to maternal gametophytic defects, atdre2-2 and atnar1-3 also have reduced male transmission, indicating a function in male gametes, whilst mutants of the CIA proteins ATR3, NPB35 and CIA1 transmitted normally through the male (Nakamura et al., 2013). Therefore, in Arabidopsis, CIA proteins share common zygotic function, while the distinct functions of AtDRE2 in female and male gametes and of AtNAR1 in male gametes are reminiscent of DEMETER hallmarks.
Further studies are needed to elucidate how AtDRE2 may control DNA methylation levels. Although Dre2 (also named CIAPIN in mouse and ANAMORSIN in yeast) has not been associated with an epigenetic role outside plants, purified CIAPIN was investigated in relation to DNA methylation. It did not show any DNA methylation or demethylation activity for DNA or RNA (Hao et al., 2008; Song et al., 2014). Two pioneering structural studies of Dre2 (Soler et al., 2012) and ANAMORSIN (Song et al., 2014) discussed putative functions of their N-terminal domains. In both proteins, the N-terminal fold belongs to the SAM methyltransferase domain superfamily (Soler et al., 2012; Song et al., 2014), characterized by the capacity to add a methyl group to a substrate, using SAM as a donor. The subtle structural differences in this N-terminal domain in Dre2 and ANAMORSIN suggest that it may be unable to bind SAM itself. Instead, it may inhibit methyl transfer by either out-competing other methyltransferases or acting as a bait for protein-protein interactions (Song et al., 2014). Interestingly, AtDRE2 genetically antagonized the maintenance DNA methyltransferase MET1 in Arabidopsis (Buzas et al., 2014). The possibility that Dre2 belongs to the SAM radical superfamily, which utilize SAM and Fe-S clusters to initiate radical reactions, was also discussed (Soler et al., 2012). The possibility that the C terminus of AtDRE2, where two Fe-S clusters can be ligated, would participate in DNA demethylation in the CC is intriguing. While AtDRE2 and DEMETER are Fe-S proteins, mutations in most genes of the Fe-S assembly pathway do not have CC and maternal seed phenotypes.
Role of CIA targeting complex proteins in DNA demethylation and repairAsymmetric leaves1/2 enhancer 7 (AE7), a protein forming a complex with the highly conserved eukaryotic CIA-targeting components CIA1, NAR1 and MET18, first came under investigation in a study about genome integrity by Luo et al. (2012). This study also suggested that ROS1 is one of the nuclear targets of the AE7-containing CIA complex.
Unlike in fungi and mammals, where MET18 is an essential protein, met18 mutants in plants are viable (Duan et al., 2015; Han et al., 2015). More than a thousand regions in met18 were differentially hypermethylated compared to wild type, and about 70% of these regions were also hypermethylated, and to a higher degree, in ros1 mutants (Duan et al., 2015). ROS1 also targets a considerably larger number of regions for 5mC BER. Moreover, taking into consideration that the Fe-S cluster binding motif is critical for ROS1 enzymatic activity, and that ROS1 directly interacts with MET18, the authors proposed a model in which MET18 is required for efficient incorporation of Fe-S clusters onto ROS1 at a subset of targets (Duan et al., 2015).
Han et al. (2015) also suggested that MET18 assembles Fe-S clusters for a small set of Fe-S proteins, in a redundant manner. First, the study confirmed the interaction between MET18 and the CIA targeting complex proteins CIA1, AE7 and NAR1. Other interactors identified were the plant catalytic subunits of DNA polymerases α, δ and ε. In yeast, these proteins have been demonstrated to contain Fe-S clusters (Netz et al., 2012). Furthermore, a number of DNA repair-related genes were up-regulated both in met18 and in viable mutants in a catalytic subunit of DNA polymerase previously implicated in DNA repair in plants (Yin et al., 2009). The authors therefore proposed that MET18 increases the efficiency of incorporation of Fe-S clusters into DNA polymerases in plants, which may also be the major targets of this protein.
In conclusion, components of the CIA targeting complex have so far been implicated in 5mC BER via ROS1 and in DNA repair via DNA polymerases. Moreover, the role of MET18 in Fe-S assembly on a subset of proteins has now been validated in plants and may be a universal feature of the CIA targeting complex.
Final remarksThe spectrum of Fe-S proteins being discovered is increasing in parallel with ongoing efforts to elucidate the roles and composition of Fe-S assembly components as well as the function of Fe-S clusters in cellular processes. Recent developments in plants may represent keys for unlocking some of the outstanding biological questions in the Fe-S cluster area in the future.
Fe-S cluster-based mechanisms appear to be widely used in DNA metabolism. The requirement for at least two enzymes that are dependent on Fe-S clusters and promote DNA demethylation, DEMETER and AtDRE2, in the central cell may indicate a need for sensing the redox environment to control removal of 5mC, especially because a temporal burst of reactive oxygen species before fertilization in the central cell has been reported (Martin et al., 2013) and coincides with DEMETER and AtDRE2 action. Nevertheless, other possibilities, for example that Fe-S clusters mediate catalysis, DNA CT or structural changes of these enzymes, are still open.
The isolation of AtDRE2 from the pFWA-GFP screen demonstrated that the central cell maternal gamete represents an experimental system where even essential components can be uncovered based on maternal seed phenotypes. The distinct maternal seed phenotype of AtDRE2 among the other CIA assembly components in Arabidopsis seems to be mirroring the unique, monothiol glutaredoxin-assisted, maturation of DRE2 in contradistinction with other Fe-S proteins known from yeast and animal studies. Therefore, in the future, the plant system may be explored to reveal requirements of maturation of AtDRE2 based on maternal seed effects.
In Arabidopsis, one pathway for maternal control of seed development is via the Fe-S dependent activity of DEMETER. Based on Fe-S assembly information, it is possible that the maternal effects are related to mitochondrial components essential for Fe-S assembly. Uncovering how the Fe-S requirement for DEMETER is fulfilled may help to answer outstanding questions in seed development.
Miyuki Nakamura is acknowledged for initial discussions on the structure and for feedback on parts of the manuscript. Hiroshi Ezura and Mariko Takayama are thanked for providing the tomato pictures. Research was supported by a Grant-in-Aid for Young Scientists B (ID 15K18558) and for Scientific Research on Innovative Areas (ID 16H01459) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.