Special reviews
Telomere biology in aging and cancer: early history and perspectives
2017 Volume 92 Issue 3 Pages 107-118

Details
2017 Volume 92 Issue 3 Pages 107-118
The ends of eukaryotic linear chromosomes are protected from undesired enzymatic activities by a nucleoprotein complex called the telomere. Expanding evidence indicates that telomeres have central functions in human aging and tumorigenesis. While it is undoubtedly important to follow current advances in telomere biology, it is also fruitful to be well informed in seminal historical studies for a comprehensive understanding of telomere biology, and for the anticipation of future directions. With this in mind, I here summarize the early history of telomere biology and current advances in the field, mostly focusing on mammalian studies relevant to aging and cancer.
All eukaryotes possess linear chromosomes, each of which must have two intrinsic ends. A natural end of a chromosome is structurally indistinguishable from an internal broken end, which causes immediate activation of the DNA damage checkpoint pathway. Once this pathway is activated, the cell cycle is halted until the damage is repaired mainly through the non-homologous end joining (NHEJ) or homologous recombination (HR) pathway. If the damage is excessive and cannot be repaired within a certain time range, the cell chooses to commit suicide (e.g., by apoptosis). Therefore, natural ends must be protected from activation of these pathways, and such a protective function is performed by a special nucleoprotein complex called the telomere. The telomere is composed of repetitive DNA, which has a 3’ protruding end called the 3’-overhang, and proteins that specifically recognize and assemble on the repetitive double- and single-stranded sequence. In mammalian systems, the protein complex is called shelterin, loss of which renders chromosome ends vulnerable to enzymatic activities, resulting in activation of the DNA damage checkpoint pathway. Loss of telomeric repeats also leads to failure of shelterin binding and end protection.
Because DNA polymerase synthesizes DNA strictly in the 5’ to 3’ orientation, the end of the lagging strand cannot be fully replicated in each S phase of the cell cycle. On the other hand, the end of the leading strand is processed to regenerate the 3’-overhang following replication. Thus, both the leading and lagging ends of replicated telomeres gradually shorten during each cell cycle unless they are replenished by a special reverse transcriptase called telomerase. Telomerase is highly expressed in unicellular eukaryotes as well as in the reproductive cells in multicellular organisms, whereas its activity is hardly detectable in normal somatic cells, which show a gradual shortening of telomeres in vitro and in vivo. Therefore, telomeres in the human soma eventually reach a critical length after a finite number of cell divisions and activate the DNA damage checkpoint, which causes irreversible cell cycle arrest (i.e., replicative senescence) and underlies cellular aging. In the absence of a functional checkpoint, cells keep dividing with ongoing telomere shortening, entering the next stage called crisis. In the crisis stage, chromosomes become unstable because of frequent end-to-end fusions, which are associated with massive cell death through mitotic catastrophe. Eventually, rare survivors that acquire the telomere maintenance pathway may emerge, resulting in tumor generation. It is also well documented that mutations in a variety of genes involved in telomere maintenance cause diseases related to human aging. Thus, telomere biology is of great importance to our understanding of human aging and tumorigenesis.
The recent technological expansion of molecular biology has greatly advanced the field, yet all of the current advances are based on seminal accomplishments by pioneers in the field. In this review, I will summarize the early history (until the first decade of this century) of telomere biology with a focus on the mammalian system to see milestone discoveries in a new light; some of these are visualized chronologically in Figs. 1 and 2. Besides the early history, recent advances in the field and perspectives will also be briefly discussed.

The history of the telomere biology, aging, and cancer fields from the early 1930s to the late 1980s. Milestone studies related to telomeric DNA and proteins (top) and aging and cancer (bottom) are aligned in chronological order.
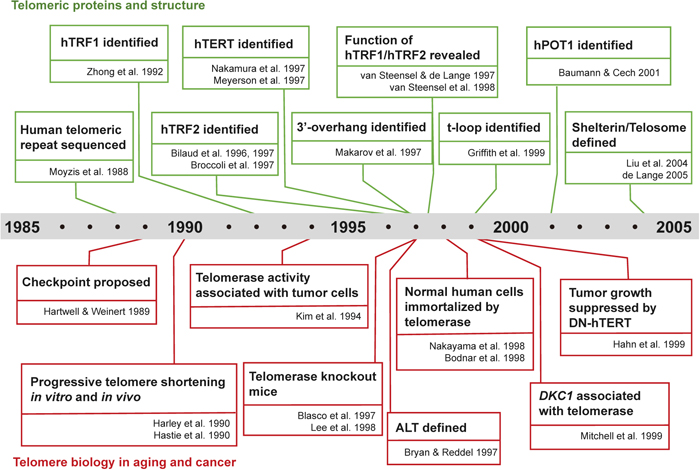
The history of telomere biology in the aging and cancer fields from the late 1980s to the early 2000s. Milestone studies related to telomeric DNA and proteins (top) and telomere biology in aging and cancer (bottom) are aligned in chronological order.
Everything has a beginning. The whole field of telomere biology was started by two Nobel Prize winners, Hermann Muller and Barbara McClintock, more than seven decades ago. Muller had studied chromosome instability, such as translocation, induced by ionizing radiation in Drosophila, which led him to find that natural chromosome ends never fuse to broken chromosome ends while broken ends often do so (Muller, 1938). He suggested the idea that natural chromosome ends are somehow protected against the repair process, which connects two broken ends, and named such a protective structure the “telomere”. A similar phenomenon was independently found by McClintock through her cytological studies using irradiated maize chromosomes (McClintock, 1931). She proposed the breakage-fusion-bridge (BFB) cycle hypothesis to explain chromosome instability: broken ends of two sister chromatids fuse together, which generates a DNA bridge during anaphase in mitosis, eventually leading to breakage of the bridge. Such broken ends will fuse again in the next cell cycle, starting a multiple fusion and breakage cycle (McClintock, 1941). She could probably have seen how relevant the BFB cycle hypothesis is in cancer biology with her own eyes in her later years.
Their seminal studies were not revisited until the 1970s, when James Watson and Alexey Olovnikov proposed the “end replication problem” (Olovnikov, 1971, 1973; Watson, 1972). They independently pointed out that the unidirectionality of DNA polymerase should leave a gap at the very end of a chromosome replicated by lagging strand synthesis. This means that telomeres should get shorter and shorter upon their replication. Such telomere shortening was proposed as the molecular basis of replicative senescence (or the Hayflick limit), an irreversible arrest of cellular growth following serial passage of cultured human cells, which was first observed by Leonard Hayflick (Hayflick and Moorhead, 1961).
Confirming this proposal at the molecular level, however, had to await the next big bang in telomere biology, which was accomplished by the 2009 Nobel Prize winners, Jack Szostak, Elizabeth Blackburn and Carol Greider. Thanks to the development of Sanger sequencing technology (Sanger and Coulson, 1975), Blackburn could successfully read telomeric sequences of the ciliated protozoan Tetrahymena thermophila (Blackburn and Gall, 1978). This finding led Szostak to devise an artificial minichromosome, capped with Tetrahymena telomeric sequences at its ends, which was successfully maintained in yeast (Szostak and Blackburn, 1982); this was the first molecular evidence that a telomeric DNA sequence possesses a protective function. The end replication problem and the structural and dynamic properties of telomeres implied the presence of enzymatic activity to counteract telomere shortening. Such an enzyme, a terminal transferase that utilizes RNA as a template, was indeed identified in Tetrahymena by Greider and Blackburn and named telomerase (Greider and Blackburn, 1985, 1987). Szostak then developed a system to identify mutants of yeast that are defective for telomere maintenance, and found that null mutants of a gene named EST1 (ever shorter telomeres) show gradual telomere shortening and a senescence phenotype (Lundblad and Szostak, 1989). This series of elegant studies pioneered a new era of telomere biology (Blackburn et al., 2006) (Fig. 1).
Telomeric DNA meets proteinAfter the telomeric DNA sequence of Tetrahymena had been published, several studies inferred the existence of telomere-binding proteins other than histones (Blackburn and Chiou, 1981; Gottschling and Cech, 1984), and such proteins were soon identified in another ciliated protozoan, Oxytricha nova (Gottschling and Zakian, 1986). Although the function of the telomeric proteins was unclear at that time, it was proposed that “they act as stability elements or chromosome caps, thereby preventing fusion of telomeres” (Gottschling and Zakian, 1986). The first evidence that a telomeric protein protects chromosome ends came from two independent studies using budding yeast, in which David Shore’s and Virginia Zakian’s groups analyzed the function of the yeast telomere-binding protein Rap1 (Conrad et al., 1990; Lustig et al., 1990).
By this time, the concept of “checkpoints” that safeguard the order of cell cycle events from intrinsic and extrinsic insults had been proposed by Leland Hartwell and Ted Weinert (Hartwell and Weinert, 1989). Among several cell cycle checkpoints, the DNA damage checkpoint is highly relevant to telomere end protection, since chromosome ends resemble DNA ends caused by DNA double-stranded breaks (DSBs), which cause a delay in DNA synthesis and were later linked to an increase of the “genome guardian”, p53 (Kastan et al., 1991). In the same year, Calvin Harley wrote that “when a certain length is reached on one or more telomeres in a dividing cell, a “checkpoint”, analogous to those regulating cell cycle events […] is signalled which evokes the Hayflick Limit: cells stop dividing” (Harley, 1991). Therefore, it was very tempting to speculate that telomeric proteins possess a protective function to prevent checkpoint activation at the chromosome ends (de Lange, 1995).
Determination of the human telomeric DNA sequence (TTAGGG)n (Moyzis et al., 1988), which is conserved among vertebrate species (Meyne et al., 1989), allowed Titia de Lange and coworkers to perform electrophoretic mobility shift assays to identify a telomere-binding activity (Zhong et al., 1992) and then clone the gene encoding a telomeric repeat binding factor, which was named TRF (for TTAGGG repeat binding factor; later renamed TRF1), from a HeLa cell extract (Chong et al., 1995). The protein was shown to be specifically localized to telomeric DNA in vivo, and overexpression of full-length and truncated dominant-negative TRF1 resulted in shortening and elongation of telomeric DNA, respectively, providing the first evidence that a mammalian telomeric protein is also involved in telomere homeostasis (van Steensel and de Lange, 1997). In the meantime, Eric Gilson’s group raised antibodies against the DNA-binding motif of a yeast telomere-binding protein, Tbf1, to search for similar proteins in human cell extracts, and identified two telomere-binding proteins (Bilaud et al., 1996), one of which turned out to be TRF1 while the other was named TRF2 (Bilaud et al., 1997). TRF2 was independently identified on the basis of its sequence similarity to TRF1 by de Lange’s group at the same time (Broccoli et al., 1997), and soon shown to be central in telomere protection: overexpression of truncated dominant-negative TRF2 caused chromosome end-to-end fusion in cultured human cells (van Steensel et al., 1998), and checkpoint kinase ATM (ataxia telangiectasia mutated)-dependent p53 activation, resulting in apoptosis or senescence in a cell type-dependent manner (Karlseder et al., 1999). These seminal studies paved the way to elucidate the molecular function of telomere-binding proteins in a variety of species. The next component of the mammalian telomeric protein complex was identified in 1999 by a yeast two-hybrid screen and named a TRF1-interacting nuclear protein, TIN2 (Kim et al., 1999). Searches for TRF2-interacting proteins by a yeast two-hybrid screen and nanoelectrospray tandem mass spectrometry independently identified Rap1, the mammalian ortholog of yeast Rap1, in the following year (Li et al., 2000; Zhu et al., 2000).
Although TRF1 and TRF2 were identified as double-stranded DNA-binding factors, the 3’ end of mammalian telomeres had been shown to possess a single-stranded TTAGGG 3’-overhang (or G-overhang) (Makarov et al., 1997), which had originally been found in hypotricha ciliates (Klobutcher et al., 1981). The telomeric protein in ciliates had been identified in 1986, as described above, which prompted biochemists to search for a similar single-stranded telomeric DNA-binding protein in higher eukaryotes. However, identification of such a protein had to await the availability of database mining. In 2001, Peter Baumann and Thomas Cech successfully identified a fission yeast single-stranded telomere-binding protein, Pot1, which has limited similarity to a portion of the telomere protein TEBP from Oxytricha and other ciliates (Baumann and Cech, 2001). A BLAST search with fission yeast Pot1 identified human POT1, which specifically binds to the single-stranded TTAGGG repeat (Baumann and Cech, 2001).
Identification of several telomeric proteins further accelerated the competition, resulting in reports of the same protein from three independent studies in 2004. de Lange and coworkers performed mass spectrometry of TRF2/TIN2-associated proteins and named a novel binding partner POT1-interacting protein 1 (PIP1) based on its ability to interact with POT1 (Ye et al., 2004); Zhou Songyang’s group took a similar approach and identified PTOP (POT1- and TIN2-organizing protein) (Liu et al., 2004b), while Susan Smith and colleagues utilized a yeast two-hybrid assay with TIN2 as a bait and reported TINT1 (TIN2 interacting protein 1) (Houghtaling et al., 2004). A single name, TPP1 (telomere protection protein 1), was given later. Biochemical analysis of the six identified proteins (TRF1, TRF2, TIN2, RAP1, POT1 and TPP1) indicated that these components form a complex on telomeric chromatin, which has been named the telosome (Liu et al., 2004a) or shelterin (de Lange, 2005), although the latter is more common in the current field.
During the period of telomere protein searches, telomeric end structure was visualized by electron microscopy, which demonstrated that telomeres can form a large duplex loop (called the t-loop) in vitro in a TRF2-dependent manner, and the t-loop can be isolated from cultured human cells (Griffith et al., 1999). The t-loop is currently the most widely accepted model for end protection, proposing the integration of the 3’-overhang into double-stranded repeats with the aid of shelterin function, especially provided by TRF2.
Telomeres are connected to aging and cancerAt the same period when replicative senescence was reported in the early 1960s, another phenomenon that is highly relevant to tumorigenesis was described: transformation of human cultured cells was characterized as the development of cytologically abnormal morphology following simian virus 40 (SV40) infection (Koprowski et al., 1962; Rabson and Kirschstein, 1962; Shein and Enders, 1962). Such transformed cells possess extended growth capacity compared to their untransformed counterparts, which is followed by massive degeneration of the transformed cultures. This is the crisis stage, and leads to almost complete loss of cellular viability; it is followed by recovery of the cultures, during which a viable minor subpopulation occasionally outgrows in the culture (Girardi et al., 1965). By the end of the 1970s, p53 had been identified as a protein exclusively expressed in transformed mouse cell lines (DeLeo et al., 1979) and co-immunoprecipitated with SV40 large-T antigen (Lane and Crawford, 1979; Linzer and Levine, 1979). Ten years later, Bert Vogelstein’s and Arnold Levine’s groups had independently reported p53’s role as a tumor suppressor (Baker et al., 1989; Finlay et al., 1989). In the same period, the first tumor suppressor gene, Rb, which causes recessive mutations that predispose cells to retinoblastoma, was identified (Friend et al., 1986), and shown to interact with the large-T antigen (DeCaprio et al., 1988). After the extensive research on replicative senescence and crisis, these two phenomena were linked to telomere biology as described briefly below (Fig. 2).
Soon after the identification of telomerase in Tetrahymena (Greider and Blackburn, 1985), human telomerase activity in HeLa cell crude extracts was reported (Morin, 1989). As predicted by the end replication problem, telomeric DNA in human cells was shown to shorten gradually during serial passage until replicative senescence in primary culture (Harley et al., 1990), as well as in vivo aging in blood samples (Hastie et al., 1990). Such gradual telomere shortening was also reported in transformed human cells until crisis, after which post-recovery outgrown cell lines were shown to have gained telomerase activity (Counter et al., 1992). Telomere shortening was well explained by the finding that no normal somatic human tissues, other than ovaries and testes, are positive for telomerase activity (Kim et al., 1994). In stark contrast, more than 90% of cultured immortal cells representing a variety of human tissues and biopsies representing many tumor types were positive for telomerase activity (Kim et al., 1994). Subsequent studies identified telomerase activity in human stem and progenitor cells, such as hematopoietic progenitor cells and T lymphocytes (Chiu et al., 1996; Weng et al., 1996), albeit not enough to maintain telomere length fully in hematopoietic stem cells (Vaziri et al., 1994). Although the underlying mechanism was still unknown, these and other studies let Harley remodel Olovnikov’s “marginotomy theory”, which predicted that critical deletion of chromosome termini eventually causes cell death (Harley, 1991; Olovnikov, 1973). The same concept was proposed by Woodring Wright and Jerry Shay as the two-stage mechanism hypothesis, in which gradual telomere shortening somehow activates the checkpoint pathway (with p53 and Rb as key factors) to induce replicative senescence, while in the absence of functional p53 and Rb, cells keep dividing with progressive telomere shortening and enter the crisis stage (Wright and Shay, 1992). Recovery from crisis by spontaneous activation of telomerase consequently gives rise to tumorigenic cells. Therefore, telomere shortening was proposed as a molecular basis of anti-tumorigenic cell cycle arrest and massive cell death in senescence and crisis, respectively. Identification of a gene encoding the human telomerase catalytic subunit (hTERT; also known as hEST2) (Meyerson et al., 1997; Nakamura et al., 1997) enabled exogenous expression of hTERT in normal human fibroblasts and epithelial cells, which resulted in extensive elongation of telomeres and immortalization of the normal cells (Bodnar et al., 1998; Nakayama et al., 1998). Ectopic expression of hTERT in SV40 large-T- and oncogenic H-ras-transformed human fibroblasts and epithelial cells resulted in bypass of crisis and tumorigenic conversion of these cells (Hahn et al., 1999a). Conversely, inhibition of telomerase activity by a dominant-negative DN-hTERT suppressed growth of human tumor-derived cell lines (Hahn et al., 1999b), demonstrating telomerase as a potential target of anti-cancer therapy. This experimental evidence established that telomere shortening underlies replicative senescence and crisis in human cultured somatic cells due to insufficient telomerase expression, and that maintenance of telomere length is essential for unlimited cellular growth.
While telomerase activity was strongly associated with tumorigenesis, there was a striking exception. A number of immortalized human cell lines are capable of elongating telomeres without any detectable telomerase activity (Kim et al., 1994; Murnane et al., 1994; Bryan et al., 1995). These cells are characterized by very long and heterogeneous telomeres, maintained by a telomerase-independent mechanism referred to as ALT (alternative lengthening of telomeres) (Bryan and Reddel, 1997), which are also found in a subset of tumor-derived cell lines and tumors (Bryan et al., 1997). Roger Reddel and coworkers showed elegantly that exogenous DNA sequences integrated into telomeres of ALT cells are copied from the original telomere to another telomere, indicating an HR-based mechanism underlying ALT (Dunham et al., 2000). Although targeting telomerase by specific inhibitors had been a promising anti-cancer drug strategy, the finding of ALT raised a significant concern for therapeutic applications of telomerase inhibitors. Because ALT-positive tumors are capable of escaping the anti-growth effect of telomeraes inhibitors, it is of great importance to develop drugs that specifically target the ALT pathway.
The fact that patients suffering from ataxia telangiectasia, which is characterized by premature aging, predisposition to cancer, immunodeficiency, hypersensitivity to ionizing radiation, and neurological disorders, show an accelerated telomere shortening and increased telomere fusion phenotype suggested a causal link between telomere maintenance and aging-related disorders (Metcalfe et al., 1996; Xia et al., 1996). In 1999, direct evidence that causally links failure of telomere maintenance and diseases of aging humans was provided. Patients with the X-linked form of a human inherited disease, dyskeratosis congenita, suffer from premature aging disorders caused by defects in highly regenerative tissues, attributable to mutations in the DKC1 gene that encodes the protein dyskerin. Kathleen Collins and colleagues revealed that dyskerin, which had previously been proposed to be involved in ribosomal function, is required for telomerase RNA generation and consequently telomerase activity and telomere maintenance (Mitchell et al., 1999). This finding was the first step to explore the causal relationship between telomere dysfunction and human degenerative disorders. Subsequently, these findings and the fact that telomeres gradually shorten in human stem cells (Vaziri et al., 1994; Lansdorp, 1998), together with mouse genetics studies described below, led to the idea that degeneration of the self-renewal capacity of stem cells is partly due to a decline of telomere length or a change in telomere structure, consequently underlying individual aging (Van Zant and Liang, 2003; Bell and Van Zant, 2004; Sharpless and DePinho, 2004). Thus, by the early 2000s, diseases associated with aging and a physiological decline accompanied by aging had both been connected to telomere biology.
What mouse genetics taught usThe telomere theory of aging and cancer was further strengthened by the development of mice lacking telomerase RNA component (mTR), which was identified in 1995 by Greider’s group (Blasco et al., 1995). Human telomerase RNA was also reported by Bryant Villeponteau and coworkers in the same year (Feng et al., 1995). In contrast to human tissues, most mouse tissues possess active telomerase (Prowse and Greider, 1995), and telomeres of laboratory-established mouse strains are kept very long (up to several hundred kb) (Kipling and Cooke, 1990). mTR–/– mice were first reported by Greider and coworkers in 1997, which unexpectedly revealed that the first few generations of knockout animals are born successfully, suggesting that telomerase is not necessary for fertility or the maintenance of renewing tissues during development (Blasco et al., 1997). However, careful analysis of later-generation animals revealed that telomere repeats are lost gradually in each generation of mice, resulting in chromosome instability, such as aneuploidy and chromosome end-to-end associations. Subsequent studies from Greider’s and Ronald DePinho’s groups revealed that telomerase is indeed required for successive maintenance of highly proliferative organs, such as testis, bone marrow and spleen, in later-generation mTR–/– mice, which show premature aging phenotypes at the organismal level, including skin lesions, alopecia and hair graying (Lee et al., 1998; Rudolph et al., 1999), supporting the notion that telomere shortening underlies the limited self-renewal capacity of stem cells in aged individuals.
Severe telomere shortening in late-generation mTR–/– mice activates the p53-dependent cell cycle arrest pathway, and loss of both mTR and p53 synergistically enhances malignant transformation (Chin et al., 1999). Such transformed cells in a p53–/– background often carry complex non-reciprocal translocations, a hallmark of the BFB cycle during tumorigenesis (Artandi et al., 2000). These seminal studies using telomerase-deficient mice gave strong support to the telomere crisis model, named by Fuyuki Ishikawa, in which the two-stage model was extended to incorporate the BFB cycle as a driver of chromosome instability and consequently tumorigenesis during the crisis stage (de Lange, 1995; Ishikawa, 1997; Artandi and DePinho, 2000).
As described above, many important findings and concepts in telomere biology had been reported by the early 2000s. Although many other telomeric accessory factors have since been identified by proteomic analysis (Déjardin and Kingston, 2009; Lee et al., 2011; Grolimund et al., 2013; Bartocci et al., 2014) and the functions of these accessory factors are important in telomere maintenance (Arnoult and Karlseder, 2015), shelterin is still recognized as the only constitutive protein complex with a direct telomere-binding ability (Doksani and de Lange, 2014). Among shelterin components that directly bind to the telomere, TRF2 has a central role in protecting telomeric ends from activating the ATM-dependent DNA damage signaling pathway and from repair through classical NHEJ, while ATR-dependent signaling is repressed by POT1 (Denchi and de Lange, 2007). Another telomere-binding protein, TRF1, is also involved in suppression of ATR signaling through promoting telomeric semi-conservative replication (Sfeir et al., 2009). The recent identification of TZAP, telomeric zinc finger-associated protein, as a TTAGGG-binding protein, however, surprised the field since it was reported to bind directly and preferentially to long double-stranded TTAGGG repeats (Li et al., 2017). Because TZAP competes with TRF2, negatively regulates telomere length and had been missed by conventional proteomics searches for shelterin-interacting proteins, it is an attractive possibility that TZAP belongs to an unknown telomeric complex.
Chromosome ends have long been thought to possess a suppressive structure against transcription because of their enrichment for heterochromatic marks (Blasco, 2007). Artificial insertion of a luciferase reporter into the subtelomere revealed 10 times less expression of the reporter compared to the same reporter in non-telomeric chromatin, suggesting that transcription is suppressed near telomeres, which has been named the telomere position effect (Baur et al., 2001). Despite this notion, a transcript that contains telomeric repeats was reported in 2007 by Joachim Lingner’s group and called telomeric repeat-containing RNA (TERRA) (Azzalin et al., 2007), proving for the first time that some subtelomeric loci are actively transcribed. Since its discovery, the TERRA non-coding transcript has been associated with proper telomere function through its interaction with various proteins, such as TRF1, TRF2, SUV39H1 and HP1 (Deng et al., 2009; López de Silanes et al., 2010; Scheibe et al., 2013; Porro et al., 2014). Interacting partners of TERRA also include telomerase RNA component, which suggests direct regulation of telomerase (Redon et al., 2010), although overexpression of TERRA does not affect telomerase activity in human cancer cells (Farnung et al., 2012). TERRA can also form a DNA:RNA hybrid with telomeric DNA, called the R-loop, which has been implicated in telomere maintenance in ALT cells by regulating telomeric recombination (Arora et al., 2014). Because of its potential telomeric function in both telomerase-positive and telomerase-negative tumors, TERRA is considered as an attractive candidate therapeutic target (Cusanelli and Chartrand, 2015).
A number of studies clearly demonstrated that the telomeric end has at least two states: a closed or capped state that protects chromosome ends from activating DNA damage checkpoints including the cell cycle arrest and damage repair pathways; and an open or uncapped state, in which both pathways are activated. Careful observation of telomere dysfunction-induced foci (TIFs), a colocalization of DNA damage marker proteins with telomeres (Takai et al., 2003), in multiple tumor cell lines indicated the existence of a third state, the intermediate state (Cesare et al., 2009) (Fig. 3). Many tumor cell lines as well as old normal human cells harbor unfused telomeres that are recognized as DNA damage, which indicates that these intermediate-state telomeres activate the DNA damage checkpoint, yet are still protected from being subjected to repair processes (Cesare and Karlseder, 2012). The intermediate-state telomere is induced by extensive telomere shortening that prevents the telomere from forming the protective closed-state structure (e.g., the t-loop), and by partial depletion of TRF2 (e.g., by shRNA-based knockdown) that leads to a failure of closed-state formation at an elongated telomere; on the other hand, the open state is a consequence of the complete loss of TRF2-binding sites, or deletion of TRF2 (e.g., by CRISPR/Cas9-based knockout) at an elongated telomere (Cesare et al., 2013). The intermediate-state telomere is proposed to induce replicative senescence without end-to-end fusion, while the open-state telomere is fused and underlies deleterious chromosome instability and cell death during the telomere crisis stage.
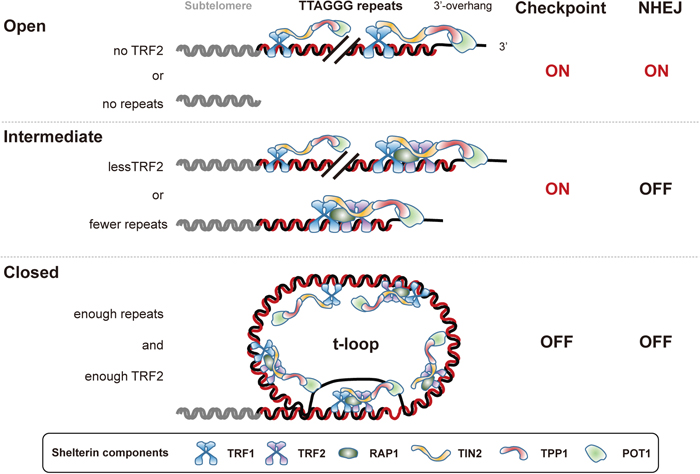
Three-state model of telomere structure. The closed state is achieved when enough telomeric repeats and TRF2 are available to form the protective t-loop structure, in which the single-stranded 3’-overhang integrates into the double-stranded repeat locus. The closed state can suppress both ATM-mediated checkpoint signaling and the classical NHEJ pathway. It is not well understood how shelterin binds to the t-loop structure. When telomeric repeats shorten or TRF2 protein declines to a level that is insufficient to support the t-loop, the telomere end state becomes intermediate. Intermediate telomeres activate the ATM checkpoint but still hold enough TRF2 to suppress NHEJ. When almost all telomeric repeats or TRF2 are removed, the chromosome end state then becomes open, which resembles an internal DSB site and activates both checkpoint signaling and the NHEJ pathway. The intermediate state is relevant to telomeres in replicative senescence, while the open state is observed in the crisis stage. Each component of shelterin is highlighted at the bottom. TRF1 and TRF2 can bind to TIN2 independently of each other, forming subcomplexes.
The intermediate-state telomere can also be induced by a prolonged arrest in mitosis (Hayashi et al., 2012). The detailed mechanism is still unclear but such mitotic telomere deprotection requires Aurora B kinase activity, suggesting an active pathway inducing the intermediate-state telomere during mitotic arrest. This partially explains a mechanism underlying cell death upon anti-tumor drug-induced mitotic arrest (Hayashi et al., 2015). The intermediate-state telomere may thus function as a biological sensor for cellular stresses, such as prolonged mitosis and cellular chronological aging (Cesare and Karlseder, 2012). What remains to be studied is whether the three-state model is relevant to tumorigenesis in vivo, and whether natural telomeres can take any additional states other than those described above.
Telomere crisisThe consequences of chromosome end-to-end fusion have been well studied in the last decade. Although recent massive genome sequencing of malignant tumor cells indicated frequent duplications in opposite orientations, which is indicative of the BFB cycle (Campbell et al., 2010; Waddell et al., 2015), live-cell analysis of epithelial cells that harbor chromosome fusions demonstrated that an anaphase chromatin bridge formed between sister nuclei persists in the next cell cycle (Pampalona et al., 2012; Maciejowski et al., 2015). A persisting chromatin bridge results in either cytokinesis failure followed by tetraploidization (Pampalona et al., 2012), or resolution by enzymatic activities (Maciejowski et al., 2015). The latter was proposed to cause kataegis, a hypermutation pattern of clustered dC>dT and dC>dG changes at TpC sites, and chromothripsis, a catastrophic rearrangement of one or a few chromosome regions in a single event (Maciejowski and de Lange, 2017). These models, including the BFB cycle hypothesis, explain how a chromosome accumulates deleterious rearrangements during telomere crisis.
On the other hand, live-cell observation of human fibroblast cells in telomere crisis indicated that cells often die during or after prolonged mitotic arrest. At least in fibroblasts, chromosome end-to-end fusion was shown to trigger mitotic arrest by an unknown mechanism, which leads to mitotic telomere deprotection and cell death during the same mitosis or the following cell cycle (Hayashi et al., 2015). Cell death following mitotic arrest is dominant in telomere crisis of fibroblasts, providing a novel insight about the mechanism of cell death during this catastrophic stage. It is worth noting that malignant tumors found in human patients are mostly of epithelial origin and fibroblast-derived sarcoma is very rare, suggesting distinct levels of resistance to tumorigenesis in these cell types. This may be explained by a strong preference for senescence in fibroblast cells compared to apoptosis in epithelial cells upon exposure to stress (Georgakopoulou et al., 2016). Another possibility that needs to be addressed, however, is that end-to-end fusion may follow a different process in each cell type, culminating in either tumorigenesis or cell death during telomere crisis.
Telomeres, aging and diseaseIt has been nearly 20 years since the first connection between telomere maintenance and dyskeratosis congenita was reported. Genetic diseases caused by malfunction of the telomere maintenance pathway have been increasingly reported and are collectively called the telomere syndromes (Armanios and Blackburn, 2012) or telomeropathies (Opresko and Shay, 2017). Mutations in genes known to function in telomere maintenance have now been identified in a variety of human genes involved in telomerase function (TR/TERC, TERT, DKC1, NOP10/NOLA3, NHP2/NOLA2, PARN, NAF1 and TCAB1/WRAP53) and telomere protection and replication (TIN2, RTEL1, POT1, CTC1, Apollo and TPP1) (Blackburn et al., 2015; Martínez and Blasco, 2017; Opresko and Shay, 2017). The cause of the telomere syndromes has been linked to the failure of stem cell replenishment, which requires a functional telomere maintenance pathway (Blackburn et al., 2015). What human telomere syndromes and mouse genetics have taught us is that telomere shortening caused by mutation in genes involved in telomere maintenance increases the risk of aging-related diseases and mortality.
Interestingly, mutations in the promoter of TERT, which increase hTERT expression only a few-fold, have been reported to cause familial and sporadic melanoma (Horn et al., 2013; Huang et al., 2013). This indicates that telomerase activity is highly regulated to be set in a certain range, otherwise the risks of aging diseases, including cancer incidence, increase. The field of telomere biology attracts public curiosity since it may have direct implications for human aging and life span. Because of this, however, careful studies will be required to dissect correlation and causality between telomere function and many aspects of human disease and health.
As briefly discussed in the last section, the field of telomere biology will undoubtedly keep expanding to fulfil the demand of public interest in human health and disease. Attaining a comprehensive understanding of the role of telomere dysfunction in aging and tumorigenesis will thus drive applied research, potentially leading to the development of effective prognostic indicators, improved therapeutic treatment, and extension of a healthy life span. As such, scientists are increasingly required to consider the impact of their work on the public. In this respect, learning how the seminal work of the pioneers has influenced current telomere biology research should help to understand the dynamics of the field in the near future. On the other hand, a number of leading studies were the result of curiosity-driven basic research, which is an important lesson from history. Some breakthroughs, such as the finding that Tetrahymena telomeres can protect the ends of minichromosome in evolutionarily distant yeast cells, have even been the result of unexpected findings. It is therefore of great importance to maintain both acknowledgement of the history of basic studies and a harmonized balance between basic and applied research for the sake of progress in science and human society.
I thank Jan Karlseder and Tomoichiro Miyoshi for critical reading of the manuscript, and Ian Smith for editing. This work is supported by grants from the Senri Life Science Foundation, the Uehara Memorial Foundation, the Daiichi Sankyo Foundation of Life Science, the Nakajima Foundation, a JSPS Grant-in-Aid for Young Scientists (A) (16H06176) and a JSPS Grant-in-Aid for Scientific Research on Innovative Areas (16H01406).