ABSTRACT
Numerous noncoding RNA transcripts are detected in eukaryotic cells. Noncoding RNAs transcribed across gene promoters are involved in the regulation of mRNA transcription via chromatin modulation. This function of noncoding RNA transcription was first demonstrated for the fission yeast fbp1 gene, where a cascade of noncoding RNA transcription events induces chromatin remodeling to facilitate transcription factor binding. We recently demonstrated that the noncoding RNAs from the fbp1 upstream region facilitate binding of the transcription activator Atf1 and thereby promote histone acetylation. Histone acetylation by histone acetyl transferases (HATs) and ATP-dependent chromatin remodelers (ADCRs) are implicated in chromatin remodeling, but the interplay between HATs and ADCRs in this process has not been fully elucidated. Here, we examine the roles played by two distinct ADCRs, Snf22 and Hrp3, and by the HAT Gcn5 in the transcriptional activation of fbp1. Snf22 and Hrp3 redundantly promote disassembly of chromatin in the fbp1 upstream region. Gcn5 critically contributes to nucleosome eviction in the absence of either Snf22 or Hrp3, presumably by recruiting Hrp3 in snf22∆ cells and Snf22 in hrp3∆ cells. Conversely, Gcn5-dependent histone H3 acetylation is impaired in snf22∆/hrp3∆ cells, suggesting that both redundant ADCRs induce recruitment of Gcn5 to the chromatin array in the fbp1 upstream region. These results reveal a previously unappreciated interplay between ADCRs and histone acetylation in which histone acetylation facilitates recruitment of ADCRs, while ADCRs are required for histone acetylation.
INTRODUCTION
Chromatin structure consisting of chromosomal DNA bound to histone-octamers plays important roles in the regulation of gene expression and genome stability. Generally, chromatin compaction inhibits many DNA-related reactions, such as transcription, by preventing the access of trans-acting DNA-binding factors to their binding sites in the DNA (Wolffe, 1997). Past studies have revealed two major classes of chromatin modifying machinery. The first class is histone modifications such as acetylation and methylation. Histone acetyltransferases (HATs) add acetyl groups that are usually associated with a derepressed chromatin state (Grant et al., 1998; Kuo and Allis, 1998). The second class of chromatin modulators is ATP-dependent chromatin remodelers (ADCRs). ADCRs alter chromatin structure by sliding or disassembling nucleosomes in an ATP-dependent manner (Travers, 1999). The yeast Swi2/Snf2 family ADCR contains a helicase/ATPase motif as a catalytic domain and a bromodomain, which recognize acetylated histone (Chandy et al., 2006; Hassan et al., 2006). Thus, proteins carrying bromodomains, including Swi2/Snf2 family ADCRs, serve as a decoder for the histone acetylation. A further class of ADCR implicated in chromatin remodeling is the CHD family ADCR that possesses two chromodomains (Travers, 1999), which recognize methylated histones (Flanagan et al., 2005). A recent study revealed the importance of this domain in enzymatic activity but not in chromatin localization of this enzyme (Morettini et al., 2011).
The Schizosaccharomyces pombe fbp1 gene has served as an excellent model system to study transcriptional regulatory mechanisms. Transcription of the fbp1 gene is regulated in response to environmental glucose (Hoffman and Winston, 1989, 1990, 1991; Vassarotti and Friesen, 1985). Under glucose starvation conditions, two critical transcription factors, Atf1 and Rst2, bind to upstream activating sequences (UAS1 and UAS2) to activate fbp1 expression (Neely and Hoffman, 2000; Janoo et al., 2001; Hirota et al., 2006). The UAS1 and UAS2 consist of a cAMP response element (CRE) and a stress response element (STRE) respectively. Moreover, the CCATT box binding factor (CBF), which includes Php5 that possesses a histone-fold domain, is required for fbp1 activation (Janoo et al., 2001). CBF binds to the UAS1-UAS2 region with a peak at UAS2 in glucose-starved cells (Asada et al., 2015). The Tup11/12 global co-repressors also bind to the fbp1 upstream region and regulate transcription to ensure a stress-specific response through chromatin regulation (Hirota et al., 2004, 2006). We also identified a cascade of long non-coding RNA (lncRNA) transcription across the fbp1 upstream region. The lncRNAs have been referred to as ‘metabolic stress-induced long non-coding RNA (mlonRNA) (Galipon et al., 2013; Miki et al., 2016), and this mlonRNA transcription is involved in the chromatin opening and the binding of transcription activators at the fbp1 promoter (Hirota et al., 2008a; Takemata et al., 2016). Our recent study demonstrated that the mlonRNAs interact with Tup11/12 corepressors to antagonize the repressive function of Tup11/12 and thereby enhance binding of Atf1, which in turn induces histone acetylation through the recruitment of Gcn5 HAT (Takemata et al., 2016). Having established the functional link between mlonRNAs, transcription activators and histone acetylation, the roles played by ADCRs in these processes remain to be elucidated. Here, we reveal a cooperative and interdependent relationship between ADCRs and histone acetylation in the chromatin remodeling for fbp1 regulation.
MATERIALS AND METHODS
Strains and media
S. pombe strains used in this study are listed in Table 1. Standard rich yeast extract medium YEL (2% glucose) was used for culturing cells (Gutz et al., 1974). YER medium (yeast extract with 6% glucose) and YED medium (yeast extract with 0.1% glucose plus 3% glycerol) was used for glucose repression and derepression, respectively (Hirota et al., 2003). All culture media used in this study contain 100 mg/L of adenine. Transformation was performed by the lithium acetate method as previously described (Hirota et al., 2001).
Table 1.
S. pombe strains used in this study
| Strain | Genotype | Reference |
|---|
| SPH1 | h− leu1-32 | (Yamada et al., 2004) |
| SPH98 | h+ ade6-M26 ura4-D18 his5-303 gcn5::ura4 | (Yamada et al., 2004) |
| SPH100 | h+ ade6-M26 snf22::ura4 ura4-D18 his5-303 | (Yamada et al., 2004) |
| SPH101 | h+ ade6-M26 snf22::ura4 gcn5::ura4 ura4-D18 his5-303 | (Yamada et al., 2004) |
| SPH102 | h+ ade6-M26 hrp1::ura4 ura4-D18 his5-303 | (Hirota et al., 2008b) |
| SPH103 | h+ ade6-M26 hrp3::ura4 ura4-D18 his5-303 | (Hirota et al., 2008b) |
| SPH105 | h+ ade6-M26 hrp3::ura4 gcn5::ura4 his5-303 ura4-D18 | (Hirota et al., 2008b) |
| SPH111 | h− ade6-M26 leu1-32 ura4-D18 fbp1(EcoT22::UAS1-2) | (Hirota et al., 2008a) |
| SPH112 | h+ ade6-M26 hrp3::ura4 snf22::ura4 his5-303 ura4-D18 | (Hirota et al., 2008b) |
| SPH116 | h− ade6-M26 leu1-32 ura4-D18 fbp1(EcoT22::nmt-terminator) | (Hirota et al., 2008a) |
| SPH243 | h− leu1-32 snf21-36 | (Yamada et al., 2008) |
| SPH331 | h− leu1-32 snf21-36 snf22::kanMX6 | This study |
| SPH332 | h+ ura4-D18 leu1-32 swr1::kanMX6 | This study |
| SPH333 | h+ ade6-M26 snf22::ura4 ura4-D18 his5-303 swr1::kanMX6 | This study |
| SPH334 | h+ ura4-D18 leu1-32 arp8::kanMX6 | This study |
| SPH335 | h+ ade6-M26 snf22::ura4 ura4-D18 his5-303 arp8::kanMX6 | This study |
The probes to detect transcripts of fbp1+ and cam1+ were prepared from PCR-amplified DNA fragments, and the DNA fragments were further labeled with 32P using a random-priming kit (GE healthcare, NJ). The nucleotide sequences of the primers used for fbp1+ and cam1+ probes were as described (Hirota et al., 2003). Total RNA was prepared from S. pombe cells by homogenizing cells in 0.3 ml of extraction buffer (0.5 M NaCl, 1% SDS, 10 mM EDTA, 0.2 M Tris-HCl [pH 8.0]), 0.3 ml of phenol-chloroform and 0.5 g of glass beads using multi beads shocker (Yasuikikai, JP). For the Northern blot analysis, 10 μg of total RNA was denatured with formamide, electrophoresed in 1.5% agarose gels containing formaldehyde and blotted to a charged Nylon membrane (Biodyne B membrane, PALL, NY).
Plasmid construction
A 1.5 kb fragment of KanMX selection marker gene (Bähler et al., 1998) was digested from pFA6a-KanMX by SmaI and EcoRI. This fragment was inserted into SmaI and EcoRI site of pBluescript KS+ to create pBS-KanMX.
Chromatin analysis and Chromatin Immunoprecipitation (ChIP)
Isolation of crude chromatin from cells and digestion with MNase were performed according to the method as described previously (Hirota et al., 2007) with slight modifications as described bellow. Fixed amount of cells (0.1 gram wet weight) were suspended in preincubation buffer (0.7 M β-mercaptoethanol, 3 mM EDTA, 20 mM Tris-HCl [pH 8.0]) and incubated at 30 ℃ for 10 min and washed once with 1 ml of ice-cold wash buffer (1.0 M sorbitol, 10 mM EDTA). Cells were centrifuged and resuspended in 0.5 ml of incubation buffer (0.75 M sorbitol, 37.5 mM Tris-HCl [pH 7.5], 1.25% Glucose, 6.25 mM EDTA). 0.5 ml of freshly prepared incubation buffer containing 2.5 mg/ml (for snf22∆ strain 5 mg/ml) of zymolyase 100T (seikagaku cooperation, JP) was added and mixed well. The cells were incubated at 30 ℃ for 5 min (for snf22∆ strain 15 min) and washed once with 1 ml of ice-cold wash buffer. Resultant spheroplasts were suspended well by pipetting in 1.4 ml of lysis buffer (18% Ficoll 400, 10 mM KH2PO4, 10 mM K2HPO4, 1 mM MgCl2, 0.25 mM EGTA, 0.25 mM EDTA, 1 mM Pefabloc SC [Roche, Mannheim, Germany]). After centrifugation at 14000 rpm for 30 min at 4 ℃, the crude nuclear pellet was resuspended in 1.5 ml of buffer A (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 5 mM KCl, 1 mM EDTA, 1 mM Pefabloc SC [Roche, Mannheim, Germany]). After addition of CaCl2 (5 mM final concentration), 0.5 ml aliquots of crude nuclear suspension were digested with different amounts of MNase (0, 20, and 50 U/ml) at 37 ℃ for 5 min. The reaction was terminated by adding 25 mM EDTA, and DNA was purified by the incubation with 1% SDS and 20 μg of proteinase K (Merck, Damstadt, Germany) at 55 ℃ followed by phenol-chloroform extraction. The DNA samples were analyzed by Southern blot as described previously (Hirota et al., 2003). Chromatin immunoprecipitation (ChIP) and quantification of ChIP DNA sample was performed as described previously (Asada et al., 2015). Following Primer sets were used in the quantitative PCR. UAS1(GGGATGAAAACAATCAACCTC and GGAATGCAGCAACGAAAATC), UAS2(GGGTGGAATGAGTCCGC and GTTCCGCGAATCATAAGCC), TATA(CGCGGAACTAAACATAGCG and GCTAGAAACCGAGTGGTG), and prp3(GCACAGTCGTTGTACAAATTCGTATTCCC, and ACGATTCTAAACGCCTCTTGTTACGATCC).
Antibodies
The following antibodies were used in this study. Anti-total histone H3 (catalog no. ab1791, Abcam, UK), Anti-H3ac (catalog no. #06-599, Millipore) and Anti-H4ac (catalog no. #06-866, Millipore).
Disruption of the fission yeast swr1 and arp8 genes
The disruption constructs for swr1 and arp8 genes, swr1-KanR and arp8-KanR were generated from genomic PCR products combined with KanMX selection marker gene. Genomic DNA sequences were amplified using the following primers for swr1: 5’-GCGGCCGCTCTAGAAGCCTCGCAACTATACTTGTCC-3’ and 5’-AGCCCGGGGGATCCACCCACTGTGAGTAACCAACAG-3’ plus 5’-TCGATACCGTCGACCGGGGCACATTGATGAGTACATG-3’ and 5’-GTACCGGGCCCCCCCCACCAATACAGAGACCAGCTTC-3’ for the left arm and right arm, respectively. Similarly, apr8 sequences were amplified using the following primers: 5’-GCGGCCGCTCTAGAACGCAATTCCCTAAAGCCGAG-3’ and 5’-AGCCCGGGGGATCCAGCGGAGTCTCGCCATATATAC-3’ plus 5’-TCGATACCGTCGACCCTGGAAGAGCGTATCCATTCC-3’ and 5’-GTACCGGGCCCCCCCGAAGGCTCCTTTGCTTCGG-3’ for the left arm and right arm, respectively. Left arm and right arm was inserted into SpeI and XhoI site of pBS-KanMX to create swr1-KanR or apr8-KanR using GENEART Seamless Cloning (Invitrogen). 5 × 107 fission yeast cells were transformed by 5 μg of swr1-KanR or arp8-KanR and spread on YE agar plate. After 16 hours, transformed cells were replica-plated on YE containing G418 (100 μg/ml) agar plate. Successful gene disruptions were confirmed by PCR using primers flanking the swr1 or arp8 genes.
RESULTS
mlonRNA transcription-mediated histone acetylation converts chromatin into an accessible state in the fbp1 upstream region
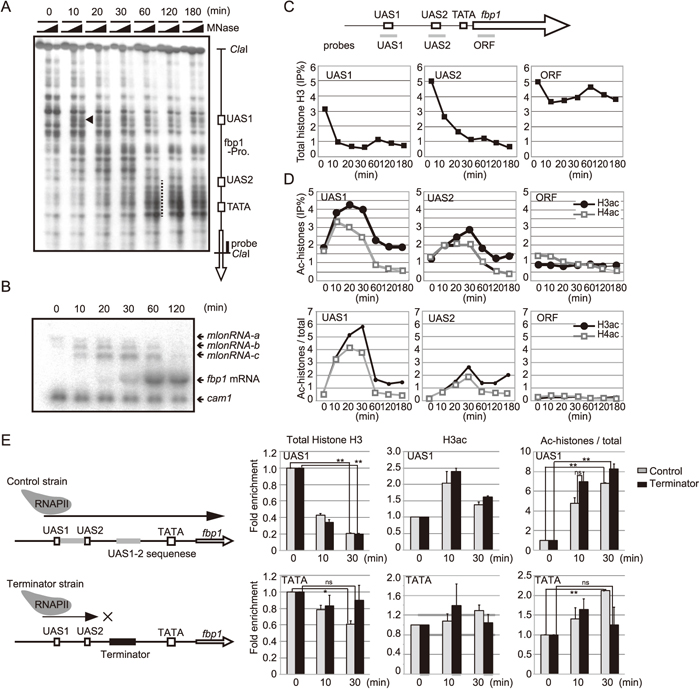
To gain insights into the link between mlonRNA transcription across the fbp1 upstream region and chromatin modulation mechanisms, we first examined the changes in the chromatin state upstream of fbp1 during glucose starvation. Micrococcal nuclease (MNase) sensitive sites reflecting an open chromatin state first appeared near UAS1 (a black arrow head in Fig. 1A), and then the chromatin around the TATA box progressively converted into an open state (a long dashed line in Fig. 1A), when massive induction of fbp1 was observed (Fig. 1A, 1B). Prior to full transcriptional activation of fbp1, long noncoding RNAs initiated from fbp1 upstream sites (referred to as mlonRNA-a, b and c) were detected as previously reported (Hirota et al., 2008a). Consistent with this observation, as well as our recent report (Takemata et al., 2016), ChIP analysis showed that histone H3 binding at UAS1 and UAS2 was decreased after glucose starvation (Fig. 1C), and acetylation levels of histone H3 and H4 peaked at 30 min after glucose starvation at UAS1 and UAS2 (Fig. 1D). Since our recent study revealed that mlonRNAs transcribed at fbp1 upstream facilitates binding of transcription factor Atf1, which is implicated in histone acetylation (Takemata et al., 2016), we assumed that the transient elevation of histone acetylation might be caused by mlonRNA transcription. To test this assumption, we inserted a transcriptional terminator sequence between UAS2 and the TATA box to arrest mlonRNAs transcription downstream of UAS2 (Fig. 1E), and measured the histone H3 acetylation at TATA box after glucose starvation. In the control cells that contain an insert of a similar size, but lacking a terminator sequence, histone acetylation was significantly increased and histone binding was decreased at 30 min after glucose starvation at both UAS1 and TATA box. On the other hand, in the terminator-inserted strain, histone acetylation and histone eviction was dramatically impaired at the TATA box, but not at UAS1 (Fig. 1E). These data indicate that mlonRNA transcription is required for histone acetylation and following disassembly of nucleosomes in this region.
Since our recent study revealed that histone H3 acetylation at fbp1 is mediated by the Gcn5 HAT (Takemata et al., 2016), we examined the role of Gcn5 in chromatin remodeling at fbp1. Additionally, we also examined the role of the Snf22 ADCR. After glucose starvation, gcn5∆ cells exhibited reduced induction of fbp1 mRNA, while snf22∆ exhibited impaired mlonRNA-c transcription and delayed expression of fbp1 mRNA, suggesting pivotal roles of Snf22 and Gcn5 in the regulation of fbp1 transcription (Fig. 2). Consistently, snf22∆ cells exhibited little chromatin alterations around UAS1 (Fig. 3A, white and gray arrows in wild-type and snf22∆ lanes, respectively) and partial chromatin opening at the fbp1 TATA box (Fig. 3A, white dashed line in snf22∆ lanes), indicating that Snf22 is involved in the chromatin remodeling at fbp1 upstream regulatory region and promoter TATA region. Interestingly, gcn5∆ cells exhibited only slightly reduced and delayed chromatin structure alterations (Fig. 3A, black dashed line in gcn5∆ lanes). Intensity of the MNase sensitive band at TATA (surrounded by white box) was quantified (Fig. 3B). These results indicate that Snf22 plays a pivotal role, while Gcn5 has a minor role, in the regulation of chromatin at fbp1. We next examined the genetic relationship between snf22 and gcn5. The snf22∆/gcn5∆ cells exhibited critically reduced fbp1 mRNA transcription and completely impaired chromatin structure alterations (Fig. 2, 3). These results indicate that Gcn5 plays a critical role in the absence of Snf22 with regard to promoter nucleosome eviction during fbp1 transcriptional activation.

We next explored the mechanisms that induce chromatin remodeling in snf22∆ cells. First, we tested the role of another class of Snf2-related ADCR, Snf21. Since Snf21 is essential for cell viability, we used a snf21-36ts mutant (Yamada et al., 2008). The snf21-36ts mutant cells exhibited slightly reduced fbp1 mRNA transcription at the restrictive temperature (37 ℃). Interestingly, snf22∆ and snf22∆/ snf21-36ts cells exhibited a similar fbp1 transcriptional defect, indicating that Snf21 and Snf22 play a role in the same pathway (Supplementary Fig. S1). Secondly, we tested the possibility that a histone variant H2A.Z plays a role in fbp1 transcriptional activation. To this end, we disrupted swr1, which encodes a histone-variant exchanger that efficiently replaces canonical histone H2A with histone H2A.Z in nucleosome arrays (Mochizuki and Yamamoto, 1992; Luk et al., 2010). The swr1∆ cells exhibited no defect in fbp1 induction upon glucose starvation (Supplementary Fig. S1). Moreover, snf22∆/swr1∆ and snf22∆ cells exhibited very similar defects in fbp1 induction (Supplementary Fig. S1). We next examined the H2A.Z removal process by Ino80 (Morrison and Shen, 2009). To this end, we disrupted arp8, which encodes a subunit of the Ino80 complex and plays critical role for this complex. The snf22∆/ arp8∆ cells and snf22∆ exhibited very similar defects in fbp1 induction (Supplementary Fig. S1). These data suggest that incorporation and removal of histone variant H2A.Z are not involved in fbp1 transcriptional control. We next analyzed the role of the Hrp3 CHD1 family ADCR, which is involved in the regulation of nucleosome positioning in fission yeast (Hennig et al., 2012; Pointner et al., 2012; Shim et al., 2012; Touat-Todeschini et al., 2012). After glucose starvation, hrp3∆ cells exhibited reduced induction of fbp1 mRNA (Fig. 2A). Moreover, the snf22∆/hrp3∆ cells exhibited critically reduced fbp1 mRNA transcription, suggesting that function of Hrp3 in fbp1 chromatin regulation is redundant with that of Snf22 (Fig. 2). Indeed, chromatin alterations at the fbp1 upstream and TATA box regions were critically impaired in the snf22∆/hrp3∆ cells (Fig. 3).
Gcn5 plays pivotal roles to induce fbp1 chromatin remodeling in hrp3∆ cells
Because both Gcn5 and Hrp3 have critical functions for chromatin remodeling in the absence of Snf22, we next investigated the genetic relationship between Hrp3 and Gcn5. Both single mutant cells exhibited mild defects in fbp1 mRNA induction upon glucose starvation (Fig. 2). Moreover, these exhibited limited defects in chromatin remodeling in the fbp1 upstream region (Fig. 3). Strikingly, hrp3∆/gcn5∆ cells exhibited little fbp1 mRNA induction and were completely impaired for chromatin remodeling (Fig. 2, 3). These data suggest that Gcn5-mediated histone acetylation might play pivotal roles in the absence of Hrp3. Taken together, concurrent loss of any two of the Snf22, Hrp3 and Gcn5 proteins results in loss of chromatin remodeling at the fbp1 upstream region and TATA box during glucose starvation (Fig. 3), suggesting a complex interplay among these factors.
Snf22 and Hrp3 are required for Gcn5-mediated histone H3 acetylation
Having established the critical role of Gcn5 in the regulation of chromatin at fbp1 in the absence of either Snf22 or Hrp3, we next asked if Gcn5-mediated acetylation is affected by Snf22 and Hrp3. To address this issue, we investigated histone acetylation at UAS1, UAS2 and the TATA box upon glucose starvation. Interestingly, in snf22∆ cells, histone H3 acetylation was partially reduced at UAS1 and critically reduced to basal levels at UAS2 and the TATA box, indicating a pivotal role of Snf22 in histone H3 acetylation (Fig. 4A). The residual histone H3 acetylation at UAS1 in snf22∆ cells was completely reduced to basal level by depleting Hrp3, suggesting that Snf22 plays a major role for histone acetylation and Hrp3 serves as a backup for Snf22.
DISCUSSION
Our recent study demonstrated the enhancer function of noncoding RNAs, which enhances transcription factor binding and thereby further induces histone acetylation as well as chromatin remodeling (Takemata et al., 2016). This current study addresses the role ADCRs to these processes. Here, we demonstrate the presence of a cooperative and interdependent relationship between two layers of chromatin modulators, ADCRs (Snf22 and Hrp3) and histone acetyltransferases.
Noncoding RNA transcription across the fbp1 upstream region induces histone acetylation at transcribed tracts
We demonstrated that histone acetylation and chromatin remodeling precede the later disassembly of the TATA box nucleosome (Fig. 1). Concurrent with the transient elevation of histone acetylation, we detected the stepwise transcription of mlonRNAs a, b and c, initiated from sites far upstream of fbp1 promoter. These observations led us to test the hypothesis that mlonRNA transcription is involved in histone acetylation. Indeed, inhibition of the mlonRNAs transcription by inserting a transcription termination sequence significantly compromised hyperacetylation of histones downstream of the termination site, but not at the upstream of this site. These data support the above hypothesis and lead us to propose a possible model for the roles of mlonRNAs to establish an open chromatin state at fbp1 (Fig. 4B, 4C). Our recent study revealed that mlonRNAs facilitate binding of Atf1 by Tup11/12-dependent and independent mechanisms (Takemata et al., 2016). In the Tup11/12 dependent mechanism, mlonRNAs interact with Tup11/12 to antagonize their repressive function and facilitate Atf1 binding, as demonstrated previously (Takemata et al., 2016). The enhanced binding of Atf1 may induce HAT recruitment to fbp1 upstream region (Fig. 4B). In the Tup11/12-independent pathway, it is possible that RNA polymerase II complexes that are transcribing mlonRNAs may play roles in enhancing DNA accessibility, as reported previously (Gilchrist et al., 2010; Weiner et al., 2010). These RNA polymerase II complexes may interact with HATs, as is the case for the interaction between PCAF HAT and RNA polymerase II (Cho et al., 1998), to induce histone acetylation over the region covered by the mlonRNA transcripts (Fig. 4C). It is possible that enhancement of Atf1 binding may be further promoted by histone acetylation following chromatin remodeling.
The cooperative and interdependent relationship between ADCRs and histone acetylation
We demonstrated that two ADCRs Snf22 and Hrp3 play a critical role for the chromatin remodeling at fbp1 and the disassembly of promoter nucleosomes. Gcn5 might facilitate both activities, because Gcn5 was critical for residual Hrp3 or Snf22 activity in snf22∆ or hrp3∆ cells, respectively. This idea is consistent with the previous findings in budding yeast that the bromodomain of Snf2 recognizes and binds to acetylated histone (Grant et al., 1998; Chandy et al., 2006; Hassan et al., 2006). The next question is how Gcn5 is involved in the regulation of Hrp3. It is possible that some proteins carrying bromodomains recruit Hrp3. Another possibility is that Gcn5 recruits and directly regulates Hrp3 through a physical interaction. This idea is supported by the finding that the budding yeast Hrp3 homolog Chd1 is part of the SAGA complex (Pray-Grant et al., 2005). The histone acetylation might be induced redundantly by Gcn5 and other HATs, since gcn5∆ cells exhibit mild defects in fbp1 mRNA induction. The NuA4 (nucleosomal acetyltransferase of histone H4) HAT (Mst1 and Mst2 in fission yeast) might be a plausible candidate, since loss of Gcn5 has a limited impact on histone H4 acetylation and concurrent loss of Gcn5 and Mst2 results in synthetic defects in the meiotic recombination at the CRE-related hotspot ade6-M26 (Hirota et al., 2008b). Conversely, we observed that Snf22 and Hrp3 are involved in the histone acetylation at fbp1 upon glucose starvation. Thus, two classes of chromatin modulators, ADCRs (Snf22 and Hrp3) and histone acetyltransferases might have a cooperative and interdependent relationship (Fig. 4D), as suggested by this study. Further understanding of the mechanism of the noncoding RNA transcription as well as the interplay between chromatin modulators will provide important clues to understand the molecular basis of chromatin regulation by noncoding RNA transcription.
ACKNOWLEDGMENTS
We acknowledge the Radioisotope Research Center in Tokyo Metropolitan University for support in the use of isotopes. This work was supported by JSPS KAKENHI (25281021, 26116518, 24114509, 16K12598, 16H02957 to KH, 26291018 to KO and 16J02252 to RA) and the Takeda Science Foundation (to KH).
CONFLICT OF INTEREST STATEMENT
There is no conflict of interest.
REFERENCES
- Asada, R., Takemata, N., Hoffman, C. S., Ohta, K., and Hirota, K. (2015) Antagonistic controls of chromatin and mRNA start site selection by Tup family corepressors and the CCAAT-binding factor. Mol. Cell Biol. 35, 847–855.
- Bähler, J., Wu, J. Q., Longtine, M. S., Shah, N. G., McKenzie, A., 3rd, Steever, A. B., Wach, A., Philippsen, P., and Pringle, J. R. (1998) Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943–951.
- Chandy, M., Gutiérrez, J. L., Prochasson, P., and Workman, J. L. (2006) SWI/SNF displaces SAGA-acetylated nucleosomes. Eukaryot. Cell 5, 1738–1747.
- Cho, H., Orphanides, G., Sun, X., Yang, X. J., Ogryzko, V., Lees, E., Nakatani, Y., and Reinberg, D. (1998) A human RNA polymerase II complex containing factors that modify chromatin structure. Mol. Cell. Biol. 18, 5355–5363.
- Flanagan, J. F., Mi, L. Z., Chruszcz, M., Cymborowski, M., Clines, K. L., Kim, Y., Minor, W., Rastinejad, F., and Khorasanizadeh, S. (2005) Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature 438, 1181–1185.
- Galipon, J., Miki, A., Oda, A., Inada, T., and Ohta, K. (2013) Stress-induced lncRNAs evade nuclear degradation and enter the translational machinery. Genes Cells 18, 353–368.
- Gilchrist, D. A., Dos Santos, G., Fargo, D. C., Xie, B., Gao, Y., Li, L., and Adelman, K. (2010) Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell 143, 540–551.
- Grant, P. A., Sterner, D. E., Duggan, L. J., Workman, J. L., and Berger, S. L. (1998) The SAGA unfolds: convergence of transcription regulators in chromatin-modifying complexes. Trends Cell Biol. 8, 193–197.
- Gutz, H., Heslot, H., Leupold, U., and Loprieno, N. (1974) Schizosaccharomyces pombe. In: Handbook of genetics. (ed.: King, R. C.), pp. 395–446. Plenum Press, New York.
- Hassan, A. H., Awad, S., and Prochasson, P. (2006) The Swi2/Snf2 bromodomain is required for the displacement of SAGA and the octamer transfer of SAGA-acetylated nucleosomes. J. Biol. Chem. 281, 18126–18134.
- Hennig, B. P., Bendrin, K., Zhou, Y., and Fischer, T. (2012) Chd1 chromatin remodelers maintain nucleosome organization and repress cryptic transcription. EMBO Rep. 13, 997–1003.
- Hirota, K., Hasemi, T., Yamada, T., Mizuno, K. I., Hoffman, C. S., Shibata, T., and Ohta, K. (2004) Fission yeast global repressors regulate the specificity of chromatin alteration in response to distinct environmental stresses. Nucleic Acids Res. 32, 855–862.
- Hirota, K., Hoffman, C. S., and Ohta, K. (2006) Reciprocal nuclear shuttling of two antagonizing Zn finger proteins modulates Tup family corepressor function to repress chromatin remodeling. Eukaryot. Cell 5, 1980–1989.
- Hirota, K., Hoffman, C. S., Shibata, T., and Ohta, K. (2003) Fission yeast Tup1-like repressors repress chromatin remodeling at the fbp1+ promoter and the ade6-M26 recombination hotspot. Genetics 165, 505–515.
- Hirota, K., Miyoshi, T., Kugou, K., Hoffman, C. S., Shibata, T., and Ohta, K. (2008a) Stepwise chromatin remodelling by a cascade of transcription initiation of non-coding RNAs. Nature 456, 130–134.
- Hirota, K., Mizuno, K., Shibata, T., and Ohta, K. (2008b) Distinct chromatin modulators regulate the formation of accessible and repressive chromatin at the fission yeast recombination hotspot ade6-M26. Mol. Biol. Cell 19, 1162–1173.
- Hirota, K., Steiner, W. W., Shibata, T., and Ohta, K. (2007) Multiple modes of chromatin configuration at natural meiotic recombination hot spots in fission yeast. Eukaryot. Cell 6, 2072–2080.
- Hirota, K., Tanaka, K., Watanabe, Y., and Yamamoto, M. (2001) Functional analysis of the C-terminal cytoplasmic region of the M-factor receptor in fission yeast. Genes Cells 6, 201–214.
- Hoffman, C. S., and Winston, F. (1989) A transcriptionally regulated expression vector for the fission yeast Schizosaccharomyces pombe. Gene 84, 473–479.
- Hoffman, C. S., and Winston, F. (1990) Isolation and characterization of mutants constitutive for expression of the fbp1 gene of Schizosaccharomyces pombe. Genetics 124, 807–816.
- Hoffman, C. S., and Winston, F. (1991) Glucose repression of transcription of the Schizosaccharomyces pombe fbp1 gene occurs by a cAMP signaling pathway. Genes Dev. 5, 561–571.
- Janoo, R. T., Neely, L. A., Braun, B. R., Whitehall, S. K., and Hoffman, C. S. (2001) Transcriptional regulators of the Schizosaccharomyces pombe fbp1 gene include two redundant Tup1p-like corepressors and the CCAAT binding factor activation complex. Genetics 157, 1205–1215.
- Kuo, M. H., and Allis, C. D. (1998) Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 20, 615–626.
- Luk, E., Ranjan, A., Fitzgerald, P. C., Mizuguchi, G., Huang, Y., Wei, D., and Wu, C. (2010) Stepwise histone replacement by SWR1 requires dual activation with histone H2A.Z and canonical nucleosome. Cell 143, 725–736.
- Miki, A., Galipon, J., Sawai, S., Inada, T., and Ohta, K. (2016) RNA decay systems enhance reciprocal switching of sense and antisense transcripts in response to glucose starvation. Genes Cells 21, 1276–1289.
- Mochizuki, N., and Yamamoto, M. (1992) Reduction in the intracellular cAMP level triggers initiation of sexual development in fission yeast. Mol. Gen. Genet. 233, 17–24.
- Morettini, S., Tribus, M., Zeilner, A., Sebald, J., Campo-Fernandez, B., Scheran, G., Wörle, H., Podhraski, V., Fyodorov, D. V., and Lusser, A. (2011) The chromodomains of CHD1 are critical for enzymatic activity but less important for chromatin localization. Nucleic Acids Res. 39, 3103–3115.
- Morrison, A. J., and Shen, X. (2009) Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nat. Rev. Mol. Cell Biol. 10, 373–384.
- Neely, L. A., and Hoffman, C. S. (2000) Protein kinase A and mitogen-activated protein kinase pathways antagonistically regulate fission yeast fbp1 transcription by employing different modes of action at two upstream activation sites. Mol. Cell. Biol. 20, 6426–6434.
- Pointner, J., Persson, J., Prasad, P., Norman-Axelsson, U., Stralfors, A., Khorosjutina, O., Krietenstein, N., Svensson, J. P., Ekwall, K., and Korber, P. (2012) CHD1 remodelers regulate nucleosome spacing in vitro and align nucleosomal arrays over gene coding regions in S. pombe. EMBO J. 31, 4388–4403.
- Pray-Grant, M. G., Daniel, J. A., Schieltz, D., Yates, J. R., 3rd, and Grant, P. A. (2005) Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature 433, 434–438.
- Shim, Y. S., Choi, Y., Kang, K., Cho, K., Oh, S., Lee, J., Grewal, S. I., and Lee, D. (2012) Hrp3 controls nucleosome positioning to suppress non-coding transcription in eu- and heterochromatin. EMBO J. 31, 4375–4387.
- Takeda, T., and Yamamoto, M. (1987) Analysis and in vivo disruption of the gene coding for calmodulin in Schizosaccharomyces pombe. Proc. Natl. Acad. Sci. USA 84, 3580–3584.
- Takemata, N., Oda, A., Yamada, T., Galipon, J., Miyoshi, T., Suzuki, Y., Sugano, S., Hoffman, C. S., Hirota, K., and Ohta, K. (2016) Local potentiation of stress-responsive genes by upstream noncoding transcription. Nucleic Acids Res. 44, 5174–5189.
- Touat-Todeschini, L., Hiriart, E., and Verdel, A. (2012) Nucleosome positioning and transcription: fission yeast CHD remodellers make their move. EMBO J 31, 4371–4372.
- Travers, A. (1999) An engine for nucleosome remodeling. Cell 96, 311–314.
- Vassarotti, A., and Friesen, J. D. (1985) Isolation of the fructose-1,6-bisphosphatase gene of the yeast Schizosaccharomyces pombe. Evidence for transcriptional regulation. J. Biol. Chem. 260, 6348–6353.
- Weiner, A., Hughes, A., Yassour, M., Rando, O. J., and Friedman, N. (2010) High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res. 20, 90–100.
- Wolffe, A. P. (1997) Histones, nucleosomes and the roles of chromatin structure in transcriptional control. Biochem. Soc. Trans. 25, 354–358.
- Yamada, K., Hirota, K., Mizuno, K., Shibata, T., and Ohta, K. (2008) Essential roles of Snf21, a Swi2/Snf2 family chromatin remodeler, in fission yeast mitosis. Genes Genet. Syst. 83, 361–372.


