Full papers
piggyBac- and phiC31 integrase-mediated transgenesis in Drosophila prolongata
2017 Volume 92 Issue 6 Pages 277-285

Details
2017 Volume 92 Issue 6 Pages 277-285
The development of transgenesis systems in non-model organisms provides a powerful tool for molecular analysis and contributes to the understanding of phenomena that are not observed in model organisms. Drosophila prolongata is a fruit fly that has unique morphology and behavior not found in other Drosophila species including D. melanogaster. In this study, we developed a phiC31 integrase-mediated transgenesis system for D. prolongata. First, using piggyBac-mediated transgenesis, 37 homozygous attP strains were established. These strains were further transformed with the nosP-Cas9 vector, which was originally designed for phiC31-mediated transgenesis in D. melanogaster. The transformation rate varied from 0% to 3.4%. Nine strains with a high transformation rate of above 2.0% were established, which will serve as host strains in future transformation experiments in D. prolongata. Our results demonstrate that genetic tools developed for D. melanogaster are applicable to D. prolongata with minimal modifications.
Development of a reliable, precise, reproducible and versatile method of transgenesis in non-model organisms is important for understanding interesting biological phenomena that are not observed in model organisms. Drosophila prolongata is a fruit fly that has unique characteristics that are not found in other Drosophila species including D. melanogaster (Setoguchi et al., 2014, 2015; Kudo et al., 2015, 2017; Hitoshi et al., 2016a). For example, males of D. prolongata have exaggerated forelegs, which are used in male–male aggressive interactions as well as in leg vibration, a unique courtship behavior observed only in this species. With these characteristics, D. prolongata serves as a good target for evolutionary studies that are not possible in D. melanogaster.
Recently, the CRISPR/Cas9 system has been introduced as a versatile tool for genome editing in non-model organisms (reviewed in Momose and Concordet, 2016; Krappmann, 2017; Taning et al., 2017). This system opened up an avenue to gene introduction into a desired site in a target genome (Salsman and Dellaire, 2017). This is advantageous compared with classical transposon-mediated transformation, in which the gene introduction site is unpredictable and the transgene is therefore vulnerable to positional effects caused by the surrounding genomic environment.
However, CRISPR/Cas9-mediated transgenesis is still insufficient for practical applications in several fields including developmental and behavioral biology. Compared with the simple marker introduction often demonstrated in the pioneering studies, transgene constructs used in contemporary developmental and behavioral biology are much larger in size, sometimes more than 15 kb, carrying various functional elements at once (e.g., Bosch et al., 2015). CRISPR/Cas9-based gene integration is not capable of introducing such large constructs (Salsman and Dellaire, 2017). Integration efficiency is another important factor to be considered for practical use. In reality, single experiments usually consist of comparisons of results obtained from multiple transgenic strains with different transgenes including positive and negative controls. Therefore, construction of each transgenic strain should be reasonably easy, otherwise tremendous effort would be required for a whole experiment to be completed. The reported gene integration efficiency of CRISPR/Cas9-mediated transgenesis in insects is much lower than would be ideal (Gratz et al., 2013, 2014; Yu et al., 2014; Gilles et al., 2015).
phiC31 integrase-mediated transgenesis is widely used in D. melanogaster, serving as a technical basis for elaborate gene manipulation in this leading model animal of genetics (Groth and Calos, 2004; Groth et al., 2004; Bateman et al., 2006; Bischof et al., 2007; Ni et al., 2009; Boy et al., 2010). It was also used for the generation of germline-specific Cas9-expressing strains, which enabled highly efficient genome editing (Kondo and Ueda, 2013). phiC31 integrase-mediated transgenesis consists of two rounds of transformation processes (Fig. 1). In the first step (phase 1), piggyBac-mediated transgenesis is used to introduce a stretch of artificial sequence into the genome (Kim and Pyykko, 2011). This sequence contains an attachment site P (attP), which serves as a target for site-specific recombination in the second step (phase 2) mediated by phiC31 integrase (Thorpe and Smith, 1998; Groth et al., 2004). Although this method requires two rounds of transgenesis and is laborious, it has many advantages compared with genome editing- and transposon-based transgenesis. First, the efficiency of the second step of transformation (phase 2) is relatively high when appropriate attP strains are used as hosts. Although the transformation rate varies among host strains, in successful cases up to 60% of fertile G0 individuals produce positive transformants in the subsequent generations (Groth et al., 2004; Bischof et al., 2007). Second, phiC31 integrase-mediated transgenesis is capable of integrating large constructs into the genome. In one report, BAC constructs of up to 133 kb were successfully introduced into the genome of D. melanogaster (Venken et al., 2006). Third, when the same host strain is used, the position of integration on the chromosome is consistent among transformants with different constructs. This feature is advantageous when the phenotype caused by different constructs is compared. Finally, excessive labor is not required once efficient host strains (attP strains) are established by the first round of transgenesis; the same attP strains are used repeatedly as a host for the introduction of different constructs.

Overview of the experimental procedures in this study. The experiment consists of a two-step transformation process. In the first step (phase 1), piggyBac-mediated transgenesis was performed to introduce an attachment site P (attP), which serves as a target for site-specific recombination in the second step (phase 2) mediated by phiC31 integrase. piggyBac transposase recognizes the piggyBac inverted terminal repeats (shown as arrows) and integrates them into the genome in a random manner. The insertion is marked by the fluorescent protein DsRed. Homozygous attP strains were established and used as host strains for phase 2 transgenesis. phiC31 integrase facilitates sequence-specific recombination between attP (inserted into chromosomes) and attB (in vectors), resulting in attL and attR. The insertion is marked with the vermilion+ gene, which is identified using adult eye color.
These features of phiC31-mediated transgenesis led us to develop a phiC31 integrase-mediated transgenesis system in D. prolongata. In this study, we established homozygous attP strains by piggyBac-mediated transgenesis. These strains were further examined for integration efficiency by phiC31-mediated transgenesis using a vector originally designed for use in D. melanogaster. Our results demonstrated that the genetic tools used in D. melanogaster are applicable to D. prolongata with minimal modifications.
Flies were maintained on ordinary cornmeal medium for Drosophila culture in a 12 h:12 h light:dark cycle (Setoguchi et al., 2014). Because development of D. prolongata is inhibited by higher temperature (Hitoshi et al., 2016b), all experiments were carried out at 20 ℃.
A vermilion− mutant was obtained by N-ethyl-N-nitrosourea (ENU) mutagenesis from an isofemale-derived strain, BaVi044 (Kudo et al., 2015). Males at the age of 4 days after eclosion were starved for 1 day by providing them with only water-soaked paper. The starved males were then fed with 5 mM ENU in 1% sucrose solution for 1 day. The ENU-fed males were transferred to a culture bottle (AS355 Drosophila stock bottle, Thermo Fisher Scientific, Waltham, MA, USA) containing cornmeal diet with 7-day-old virgin females (20 males and 20 females in a bottle) and allowed to mate (G0). The flies were transferred to a new bottle every 2 days, four times, producing five bottles of culture in 10 days. The females of the next generation (G1) were individually crossed with three males each, and a vermilion− mutant fly was found among the males of the next generation (G2). The mutant fly was crossed with wild-type females, and a homozygous line was established by crossing the individuals of subsequent generations. The mutation was confirmed to be on the sex chromosome by diagnostic crosses. The genomic sequence corresponding to the vermilion− ORF was sequenced and a nucleotide substitution, A1070G, which caused the amino acid substitution L358R, was found in the mutant strain. This mutation substitutes a hydrophobic residue at a site that is highly conserved among arthropods with a basic residue, and is presumed to have abolished the protein’s function.
Construction of vectors and helper plasmidspBac[3xP3DsRed-attP(AF)] was generated by cloning a 161-bp attP fragment, amplified by PCR from piggyBac-yellow+-3-attP (Venken et al., 2006) using the primer pair 5’-AAGGAAAAAAGGCGCGCCCTTCACGTTTTCCCAGGTC-3’ and 5’-TCCCGGCCGGCCTCGCGCTCGCGCGACTGACG-3’, into AscI- and FseI-digested pBac[tetO-EGFP, 3xP3-DsRed] (Hara et al., 2008), which is equivalent to pBac[3xP3-DsRedaf] (Horn et al., 2002) (Fig. 2). pSP64_poly(A)/piggyBac_ORF helper plasmid was generated by cloning the 1.8-kb piggyBac transposase ORF fragment, amplified by PCR from phsp-pBac (Handler and Harrell, 1999) using the primer pair 5’-CCTTGTCGACTATGGGATGTTCTTTAGACGA-3’ and 5’-CCAAGGATCCTTTAGTCAGTCCAGAAACAAC-3’, into SalI- and BamHI-digested pSP64_poly(A) vector (Promega, Madison, WI, USA). pBFv-nosP-Cas9 (Kondo and Ueda, 2013) was used without modification. pET11phiC31poly(A) was a gift from Dr. Michele Calos (Addgene plasmid # 18942; Groth et al., 2004).

Structure of pBac[3xP3DsRed-attP(AF)]. pBac-ITR: piggyBac inverted terminal repeat sequence; 3xP3-hsp70: three tandem repeats of Pax-6/eyeless binding elements followed by a minimal promoter from hsp70.
The vector plasmids pBac[3xP3DsRed-attP(AF)] and pBFv-nosP-Cas9 were purified using a QIAGEN Plasmid Mini kit (Qiagen, Hilden, Germany). piggyBac transposase mRNA was in vitro-transcribed using a mMESSAGE mMACHINE SP6 kit (Thermo Fisher Scientific) from EcoRI-linearized pSP64_poly(A)/piggyBac_ORF. The phiC31 integrase mRNA was transcribed using a mMESSAGE mMACHINE T7 Ultra kit (Thermo Fisher Scientific) from BamHI-linearized pET11phiC31poly(A). Both mRNAs were purified using a MEGAclear kit (Thermo Fisher Scientific).
The vector DNA and helper mRNA were mixed and diluted in distilled water at the following concentrations. For the piggyBac-based transgenesis (phase 1), two concentrations (300 and 500 ng/μl) of the vector DNA (pBac[3xP3DsRed-attP(AF)]) were tested with a fixed concentration of the piggyBac transposase mRNA (300 ng/μl). For phiC31 integrase-mediated transgenesis (phase 2), concentrations of the vector DNA (pBFv-nosP-Cas9) and the phiC31 integrase mRNA were 600 and 200 ng/μl, respectively.
MicroinjectionEggs were collected from a population comprising approximately 100 sexually mature flies (7–15 days old, female:male = 1:1) in an empty culture bottle. Females were allowed to lay eggs for 1 h on a petri dish (35 mm diameter) filled with instant Drosophila medium (Formula 4–24 Blue, Carolina Biological Supply Co., Burlington, SC, USA) supplemented with an equal amount of glucose and a small amount of dry yeast. Using forceps, the eggs were removed from the medium, rinsed with water and placed on a slide glass with double-sided tape on the surface. After several minutes for desiccation, the eggs were dechorionated using forceps and aligned on another slide glass treated with glue extracted from double-sided tape (Scotch W-18, 3M Japan, Tokyo, Japan) by heptane. After several minutes of desiccation (the length is critical and is determined depending on ambient humidity each day), the eggs were covered with silicone oil (FL-100-1000CS, Shin-Etsu Chemical Co., Tokyo, Japan). The microinjections were performed using an inverted phase contrast microscope (Primo vert, Carl Zeiss Microscopy, Jena, Germany) with Femtotips II (Eppendorf, Hamburg, Germany) attached to a joystick manipulator (MN-151, Narishige, Tokyo, Japan). The injected eggs on the slide glass were placed in a humidified box for 2 days, and hatched larvae were collected and transferred into a vial containing cornmeal medium.
Crossing and verification of transformantsFor piggyBac-mediated transgenesis (phase 1), the vermilion− mutant was used as the host strain. G0 virgin adults were crossed individually with three vermilion− flies of the opposite sex (Fig. 3). G1 larvae were screened for red fluorescence using a fluorescence stereomicroscope (MZ-FL III, Leica Microsystems, Wetzler, Germany), and fluorescence-positive transgenic larvae were transferred to a new vial. Virgin G1 adults were crossed individually with three vermilion− flies of the opposite sex. Up to five G1 crosses were made for each G0 family. G2 larvae were screened for red fluorescence and positive larvae were transferred to a new vial. Emerged G2 transgenic individuals derived from the same G1 individuals were crossed with each other, and homozygous G3 larvae were screened according to the intensity of fluorescence. G3 adults were crossed as independent pairs of single males and females (up to five pairs for each G1 family when homozygous G3 larvae were unambiguously distinguished by fluorescence; if not, up to 25 pairs). Homozygosity was examined by crossing at least three G4 virgin females individually with vermilion− males. When all G5 larvae were fluorescence-positive, the corresponding G4 stock was considered as homozygous for attP insertion.
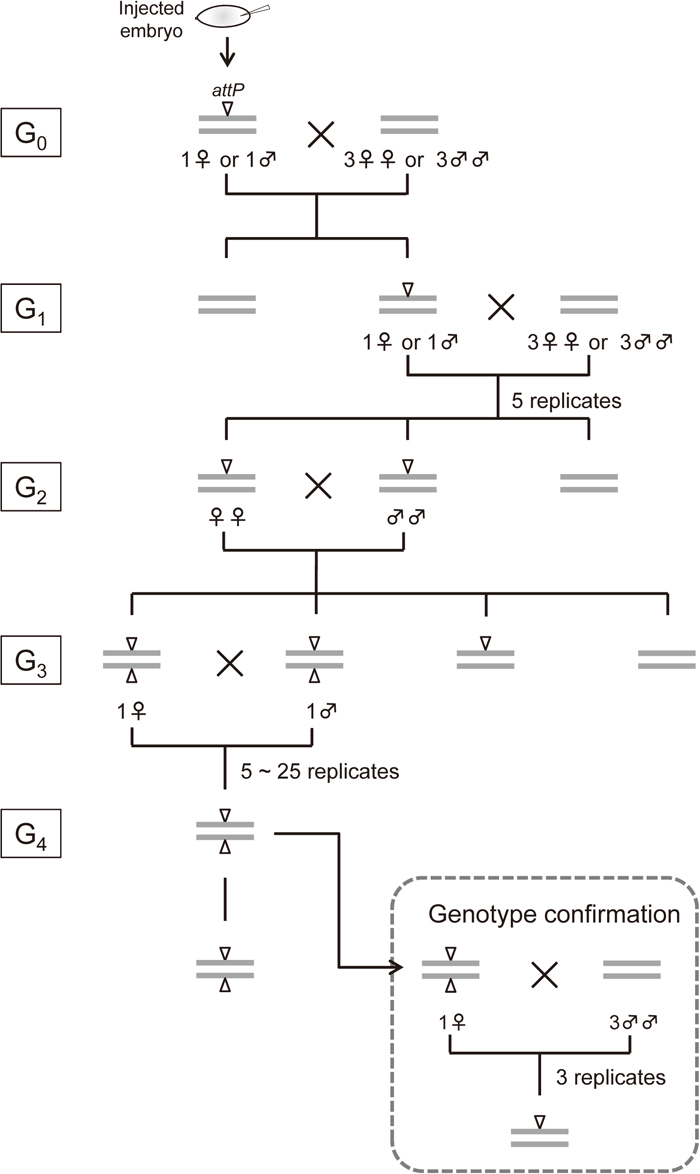
Crossing scheme of piggyBac-mediated transgenesis (phase 1). Gray bars represent the genome (for G0, the genome in germline cells) and arrowheads represent attP insertion. Only a single pair of autosomes is shown for simplicity. G0 adults derived from microinjected embryos were crossed individually to obtain G1 larvae, which were screened for DsRed fluorescent marker expression. For each G0 family, five G1 adults were individually crossed to obtain G2 larvae, which were again selected for DsRed expression. G2 adults derived from the same G1 parents were crossed with each other. G3 larvae that were homozygous for each insertion were identified using the intensity of DsRed expression, which was higher in homozygotes than in heterozygotes. G3 adults were crossed as independent pairs of single males and females (up to five pairs when homozygotes were unambiguously distinguished; otherwise up to 25 pairs). G4 families were established as homozygous attP strains, whose genotype was confirmed by three replicates of diagnostic crosses.
For phiC31 integrase-mediated transgenesis (phase 2), the homozygous attP strains generated in phase 1 were used as host strains. For each attP strain, at least 180 embryos were microinjected. G0 virgin adults were crossed individually with three vermilion− flies of the opposite sex (Fig. 4). G1 adults were screened for eye color, and vermilion+ individuals were considered as transformants.

Crossing scheme of phiC31-mediated transgenesis (phase 2). Filled arrowheads represent a nosP-Cas9 construct integrated into the attP site, which was identified by adult eye color rescued by the vermilion+ marker gene.
In order to use a visible marker in phiC31-mediated transgenesis (phase 2), we first generated a vermilion− mutant strain, by ENU mutagenesis, which was functionally rescued by the vermilion+ gene of D. melanogaster (see next section). Eggs of the vermilion− strain showed high hatchability, which was approximately 70% for dechorionated, non-microinjected embryos (data not shown). Two concentrations of the vector DNA were tested, and hatchability was approximately 50% for both concentrations (Table 1). Approximately half of the hatched larvae survived until the adult stage, and two-thirds of these were fertile. Transformation efficiency among the fertile G0 crosses was different between the two vector concentrations: twice the number of germline transformants was obtained with the higher concentration of DNA from the same number of fertile G0 crosses.
| Experiment | Vector/helper concentration (ng/μl) | Injected embryos | Hatched larvae | G0 adults | Fertile G0 | Transformed G0† | Transformation efficiency (%)‡ | Transformation rate (%)§ |
|---|---|---|---|---|---|---|---|---|
| 1 | 300/300 | 764 | 351 (45.9%) | 159 (45.3%) | 110 (69.2%) | 5 | 4.5 | 0.7 |
| 2 | 500/300 | 1264 | 805 (63.7%) | 465 (57.8%) | 362 (77.8%) | 39 | 10.8 | 3.1 |
Each transformant was further crossed to establish a homozygous strain (Fig. 3). Because a single G0 individual may carry multiple (independent) piggyBac insertions, we first crossed these individuals with vermilion− flies, and the resulting G1 transformants were again crossed individually with vermilion− flies. When G2 larvae derived from the same G0 individual showed different DsRed expression patterns, they were treated as independent strains. At this step, a total of 56 heterozygous strains were established. Each strain was named after the G0 serial number. Ten strains (attP223, att243, attP252, attP403, attP415, attP418, attP429, attP506, attP589 and attP602) that produced heterogeneous DsRed expression at the G1 generation were further divided into two lines with suffix A or B. Although we did not exclude possible insertions into the sex chromosome from our crosses, all of the obtained attP insertions were on autosomes. Establishing homozygous stocks was laborious because no balancer chromosomes or chromosome markers were available. In some strains, homozygotes could be identified by a higher intensity of DsRed fluorescence than heterozygotes. In total, 412 G3 crosses and 620 G4 diagnostic crosses were made, and more than 6,800 larvae were examined for fluorescence. As a result, 37 homozygous attP strains were established (Table 2). These strains have been stably maintained for more than 20 generations.
| Strain | Injected embryos | Hatched larvae | G0 adults | Fertile G0 | Transformed G0† | Transformation efficiency (%)‡ | Transformation rate (%)§ |
|---|---|---|---|---|---|---|---|
| attP35 | 199 | 105 (52.8%) | 55 (52.4%) | 27 (49.1%) | 2 | 7.4 | 1.0 |
| attP42 | 182 | 79 (43.4%) | 43 (54.4%) | 31 (72.1%) | 1 | 3.2 | 0.5 |
| attP54 | 197 | 136 (69.0%) | 92 (67.6%) | 55 (59.8%) | 3 | 5.5 | 1.5 |
| attP55 | 197 | 124 (62.9%) | 69 (55.6%) | 35 (50.7%) | 1 | 2.9 | 0.5 |
| attP87 | 210 | 108 (51.4%) | 75 (69.4%) | 55 (73.3%) | 2 | 3.6 | 1.0 |
| attP91 | 194 | 112 (57.7%) | 64 (57.1%) | 49 (76.6%) | 3 | 6.1 | 1.5 |
| attP178 | 198 | 146 (73.7%) | 92 (63.0%) | 73 (79.3%) | 0 | 0.0 | 0.0 |
| attP223 | 191 | 114 (59.7%) | 61 (53.5%) | 34 (55.7%) | 0 | 0.0 | 0.0 |
| attP243 | 202 | 111 (55.0%) | 65 (58.6%) | 52 (80.0%) | 5 | 9.6 | 2.5 |
| attP252A | 205 | 131 (63.9%) | 87 (66.4%) | 68 (78.2%) | 0 | 0.0 | 0.0 |
| attP252B | 197 | 129 (65.5%) | 88 (68.2%) | 49 (55.7%) | 4 | 8.2 | 2.0 |
| attP403B | 201 | 132 (65.7%) | 69 (52.3%) | 33 (47.8%) | 2 | 6.1 | 1.0 |
| attP405 | 199 | 132 (66.3%) | 91 (68.9%) | 73 (80.2%) | 2 | 2.7 | 1.0 |
| attP413 | 198 | 139 (70.2%) | 92 (66.2%) | 73 (79.3%) | 3 | 4.1 | 1.5 |
| attP415A | 194 | 122 (62.9%) | 80 (65.6%) | 45 (56.3%) | 0 | 0.0 | 0.0 |
| attP415B | 194 | 122 (62.9%) | 77 (63.1%) | 48 (62.3%) | 1 | 2.1 | 0.5 |
| attP420 | 196 | 117 (59.7%) | 74 (63.2%) | 58 (78.4%) | 5 | 8.6 | 2.6 |
| attP429A | 202 | 130 (64.4%) | 79 (60.8%) | 55 (69.6%) | 6 | 10.9 | 3.0 |
| attP429B | 213 | 148 (69.5%) | 81 (54.7%) | 42 (51.9%) | 0 | 0.0 | 0.0 |
| attP440 | 194 | 127 (65.5%) | 93 (73.2%) | 61 (65.6%) | 5 | 8.2 | 2.6 |
| attP441 | 202 | 131 (64.9%) | 54 (41.2%) | 37 (68.5%) | 0 | 0.0 | 0.0 |
| attP446 | 197 | 114 (57.9%) | 87 (76.3%) | 64 (73.6%) | 3 | 4.7 | 1.5 |
| attP449 | 204 | 124 (60.8%) | 75 (60.5%) | 44 (58.7%) | 7 | 15.9 | 3.4 |
| attP460 | 197 | 112 (56.9%) | 76 (67.9%) | 42 (55.3%) | 5 | 11.9 | 2.5 |
| attP490 | 199 | 121 (60.8%) | 75 (62.0%) | 44 (58.7%) | 1 | 2.3 | 0.5 |
| attP506 | 190 | 110 (57.9%) | 65 (59.1%) | 46 (70.8%) | 4 | 8.7 | 2.1 |
| attP525 | 199 | 124 (62.3%) | 80 (64.5%) | 71 (88.8%) | 3 | 4.2 | 1.5 |
| attP539 | 196 | 137 (69.9%) | 69 (50.4%) | 40 (58.0%) | 5 | 12.5 | 2.6 |
| attP557 | 209 | 138 (66.0%) | 78 (56.5%) | 57 (73.1%) | 2 | 3.5 | 1.0 |
| attP566 | 210 | 123 (58.6%) | 75 (61.0%) | 54 (72.0%) | 5 | 9.3 | 2.4 |
| attP589A | 199 | 132 (66.3%) | 94 (71.2%) | 58 (61.7%) | 0 | 0.0 | 0.0 |
| attP589B | 194 | 133 (68.6%) | 103 (77.4%) | 60 (58.3%) | 1 | 1.7 | 0.5 |
| attP602B | 196 | 145 (74.0%) | 67 (46.2%) | 55 (82.1%) | 0 | 0.0 | 0.0 |
| attP623 | 196 | 110 (56.1%) | 71 (64.5%) | 53 (74.6%) | 2 | 3.8 | 1.0 |
| attP634 | 199 | 143 (71.9%) | 83 (58.0%) | 59 (71.1%) | 3 | 5.1 | 1.5 |
| attP635 | 196 | 121 (61.7%) | 69 (57.0%) | 44 (63.8%) | 1 | 2.3 | 0.5 |
| attP636 | 198 | 98 (49.5%) | 41 (41.8%) | 15 (36.6%) | 1 | 6.7 | 0.5 |
To demonstrate phiC31 integrase-mediated transgenesis in D. prolongata, we selected the nosP-Cas9 construct originally developed for use in D. melanogaster, mainly because it carries the vermilion+ gene as a visual marker and was readily available for this experiment. In addition, the resulting transformants were expected to express Cas9 protein in germline cells under the control of the nanos promoter, and these will be used in prospective gene editing experiments. Beyond the demonstration of transgenesis itself, the phase 2 experiment also aimed to screen the attP strains for their transgenic efficiency. Because each attP strain should have different gene integration efficiency, it is important to identify which attP strains have a high enough efficiency for practical use. To expect at least one transformant among 100 microinjected embryos in 95% of experiments, the transformation rate should be higher than 3%. To achieve this goal, approximately 200 embryos were microinjected with the nosP-Cas9 construct for each attP strain, and the proportion of G0 individuals that produced transgenic G1 individuals was calculated (Table 2). As a result, we obtained one strain with a transformation rate of more than 3% and eight additional strains with rates above 2%.
The ENU-induced vermilion− mutant in D. prolongata was functionally rescued by the vermilion+ gene of D. melanogaster, showing that 1) the L358R substitution in the vermilion gene of the D. prolongata mutant abolished the protein function as expected, and 2) the wild-type vermilion gene function is conserved between the two species. As expected from the relatively close phylogenetic relationship between D. prolongata and D. melanogaster, the function of other genes could also be conserved between the two species, which is advantageous when vectors originally designed for D. melanogaster are to be used in D. prolongata.
Delivery of the enzymes as mRNAThe enzymes required for transgenesis (transposase and integrase) are often delivered as DNA in the form of a helper plasmid, mainly because of the ease of preparation. However, some problems have been reported with this method. For example, helper plasmids are more toxic than mRNAs (Shinmyo et al., 2004). In addition, the promoter needs to be compatible with the cellular transcription machinery for efficient expression of the enzymes; therefore, the risk of promotor incompatibility needs to be taken into account when the helper vector is used in species other than the one for which it was originally designed. In contrast, mRNA application is free from the transcription problem. In this study, piggyBac transposase and phiC31 integrase were supplied as mRNAs, and satisfactory results were achieved. Although the preparation and handling of mRNAs are costly and laborious, we adopted this method to avoid potential problems associated with the DNA method. As a result, the total transformation rate was as high as 2.2% in the phase 1 experiment; this was higher than most reported values for piggyBac-mediated transgenesis in other Drosophila species: 0.2–0.6% in D. melanogaster, 0.7% in D. willistoni, and 5.3% in D. suzukii (Handler and Harrell, 1999, 2001; Finokiet et al., 2007; Schetelig and Handler, 2013). The good performance in D. prolongata is attributable at least in part to the high viability and fertility of the microinjected G0 individuals in this species.
Variable transformation efficiency among the attP strainsBecause the expression pattern of transgenes is known to be affected by the genomic environment surrounding the insertion site (positional effect), in a practical application, possible positional effects are usually discriminated by comparing the results obtained from different attP strains that were transformed with the same construct (Bischof et al., 2007; Stern et al., 2017). Therefore, multiple attP strains should be established for practical use. Preferably, the integration efficiency of these attP lines should be considerably high because they will be used repeatedly for various experiments. To meet these prerequisites, as many attP strains as possible need to be generated at the first round of transgenesis (phase 1). We generated 56 attP strains, of which 37 were established as homozygous strains. The transformation rate among these strains varied from 0% to 3.4%.
The major reason for this variation could be the different chromatin environment around each attP site, which may prevent physical access to the attP site in some cases. However, other factors may also affect the transformation rate. For example, the quality of the co-injected phiC31 integrase mRNA might influence the integration efficiency. Because microinjection was carried out on different days using multiple batches of in vitro-transcribed mRNA, we cannot completely exclude this possibility. Nevertheless, it should be noted here that no batches of mRNA failed to generate any transformants, and every batch showed variation in the transformation rate depending on the host strains, suggesting that the effect of the mRNA quality was limited. The condition of parental flies from which the embryos for microinjection were collected might also affect the transformation rate via the hatchability of embryos. This effect was minimized by repeating microinjection into the same strain on several different dates. Although these factors might reduce the observed transformation rate, they would not raise it. Therefore, our conclusion on the strains with high rates is not affected by the above factors and these strains are considered to be usable for prospective applications.
We thank T. Niimi, H. Bellen, and the National Institute of Genetics Fly Stock Center for DNA constructs; and U. Umeki for technical assistance. This work was supported by Japan Society for the Promotion of Science grant #26660263 to T. M. The authors have no conflict of interest.