ABSTRACT
N6-methyladenosine (m6A) modifications are the most abundant internal modifications of mRNA and have a significant role in various cancers; however, the m6A methylome profile of oral squamous cell carcinoma (OSCC) in the mRNA-wide remains unknown. In this study, we examined the relationship between m6A and OSCC. Four pairs of OSCC and adjacent normal tissues were compared by Methylated RNA immunoprecipitation sequencing (MeRIP-seq). Gene Ontology, Kyoto Encyclopedia of Genes and Genomes (KEGG), and Ingenuity Pathway Analysis (IPA) analyses were used to further analyze the MeRIP-seq data. A total of 2,348 different m6A peaks were identified in the OSCC group, including 85 m6A upregulated peaks and 2,263 m6A downregulated peaks. Differentially methylated m6A binding sites were enriched in the coding sequence in proximity to the stop codon of both groups. KEGG analysis revealed genes with upregulated m6A-modified sites in the OSCC group, which were prominently associated with the forkhead box O (FOXO) signaling pathway. Genes containing downregulated m6A-modified sites were significantly correlated with the PI3K/Akt signaling pathway, spliceosome, protein processing in the endoplasmic reticulum, and endocytosis. IPA analysis indicated that several genes with differential methylation peaks form networks with m6A regulators. Overall, this study established the mRNA-wide m6A map for human OSCC and indicated the potential links between OSCC and N6-methyladenosine modification.
INTRODUCTION
Oral squamous cell carcinoma (OSCC) is the most common malignant tumor of the oral cavity and affects millions of people worldwide (Reyes-Gibby et al., 2014; Lee et al., 2017). A considerable number of OSCC patients usually undergo complicated surgery with a poor prognosis (Dave et al., 2020). Despite advances in recent decades, the overall survival rate of patients with OSCC remains low (about 50%) without significant progress (Mascitti et al., 2018; Cristaldi et al., 2019). Therefore, to find new therapeutic targets, identifying the molecular mechanism underlying OSCC is necessary.
N6-methyladenosine (m6A) modification is the most abundant internal mRNA modification and is closely linked with tumorigenesis (Du et al., 2022), proliferation (Zhang et al., 2022), invasion (Cheng et al., 2019), and metastasis (Liao et al., 2022). The essential role of m6A modification in OSCC was revealed in several studies. METTL3 (methyltransferase-like 3) increases the stability of IGF2BP2-mediated SLC7A11 mRNA, which promoting OSCC progression (Xu et al., 2021). Heterogeneous nuclear ribonucleoprotein A2B1 (hnRNPA2B1) identifies m6A motifs (Sun et al., 2019) and participates in the subcellular localization and translation of mRNA (Dreyfuss et al., 2002). However, the m6A methylome of mRNA in OSCC remains uncertain. High-throughput experimental methods, such as Methylated RNA immunoprecipitation sequencing (MeRIP-seq), has been used for identifying RNA methylation sites (Dominissini et al., 2012; Meyer et al., 2012). Recently, many computational prediction models of m6A modification have been proposed, such as BERT6mA and CNN6mA. Compared with traditional technologies, they have the advantages of reduced time, low cost, and a high prediction accuracy; thus, they are expected to be widely adopted in the future (Tsukiyama et al., 2022, 2023).
In the present study, we examined m6A modification in OSCC and determined the potential relevance to provide insight into the underlying mechanisms. Figure 1 showed the analysis workflow for this study. Using MeRIP-seq analysis, differential methylation peaks were identified in mRNA by comparing OSCC samples with normal adjacent tissues and the basic characteristics of mRNA-wide m6A modifications in human OSCC were determined. The differential m6A peak genes were analyzed by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analysis to determine the mechanisms or pathways by which m6A modification is associated with OSCC. Moreover, m6A regulators and selected genes (the genes selected from the most prominently enriched pathways) with differential methylation peaks were imported into the Ingenuity Pathway Analysis (IPA) platform to examine their direct or indirect connection to provide insight for further study.
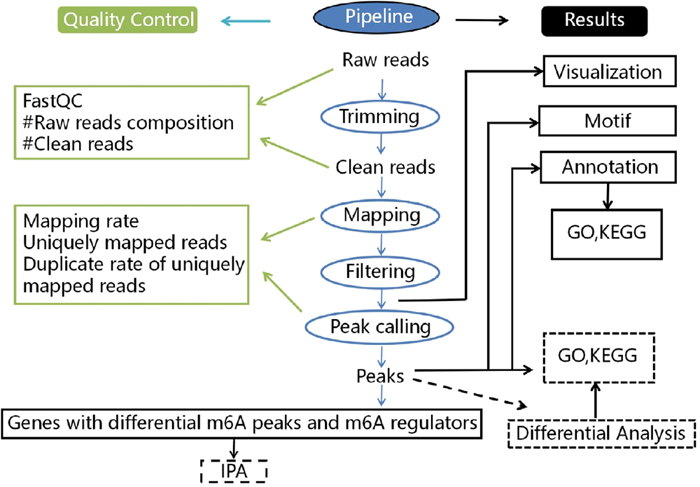
Fig. 1. Work flow chart for this study.
RESULTS
Annotation characteristics of m6A peak modification in OSCC samples and adjacent normal tissues
We used FastQC software to conduct basic quality control on the raw data and the results (Supplementary Fig. S1A–S1G) to determine whether they met our experimental requirements. MeRIP-seq analysis was performed on OSCC samples and normal adjacent tissues from four OSCC patients. We detected 20,298 m6A peaks in these two groups of samples. Compared with normal tissues, 2,348 m6A peaks were different in OSCC tissues (Fig. 2A), including 85 upregulated m6A peaks and 2,263 downregulated m6A peaks (Fig. 2B). In both OSCC and adjacent normal tissues, m6A peaks were primarily enriched within the coding sequence in proximity to the stop codon (Fig. 2C–2E). Nonetheless, m6A peaks in the OSCC tissues exhibited a different pattern from the peaks in adjacent normal tissues. Compared with normal samples, the number of m6A peaks in OSCC samples was increased in the 5’UTR (3.48% vs 2.72%) and CDS (45% vs 2.72%), and decreased in the 3’UTR (38.43% vs 40.09%) (Fig. 2D–2E). Next, we determined the distribution of m6A peaks on chromosomes in both groups. As shown in Fig. 2F–2G, dysregulated N6-methyladenosine peaks were found in all chromosomes, except the Y chromosome, but particularly in chromosomes 1–5 and the X chromosome. In addition, m6A motifs were identified in mRNA regions where m6A methylation occurred as determined by HOMER software (Fig. 2H–2K).
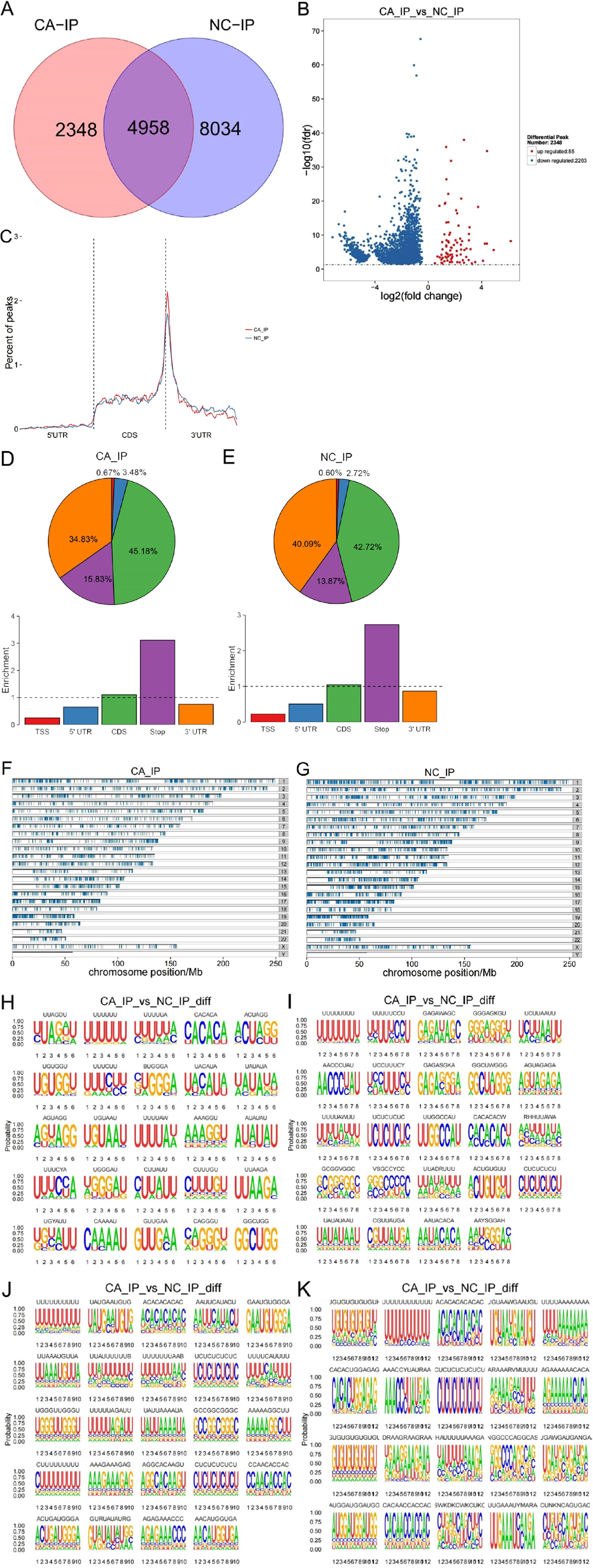
Fig. 2. Characteristics of m6A methylation in oral squamous cell carcinoma (OSCC). (A) Detected m6A peaks in OSCC samples and adjacent normal tissues. (B) Different m6A peaks between OSCC and adjacent normal groups. (C) The location distribution relative to genes of the m6A peaks in OSCC and adjacent normal groups. (D) The distribution of m6A peaks on the functional region of the mRNA transcript in CA_Input. (E) The distribution of m6A peaks on the functional region of the mRNA transcript in NC_Input. (F–G) The distribution of m6A peaks on chromosomes in CA_Input and NC_Input. When the number of chromosomes containing peaks was greater than 25, only the distribution of peak in 25 chromosomes is displayed. (H) The most significant 25 motifs with a length of 6 between the OSCC and adjacent normal groups. (I) The most significant 25 motifs with a length of 8 between the OSCC and adjacent normal groups. (J) The most significant 25 motifs with a length of 10 between the OSCC and adjacent normal groups. (K) The most significant 25 motifs with a length of 12 between the OSCC and adjacent normal groups.
A pod diagram was used to observe the overall distribution of log10 read density (RPM) of differential m6A peaks in the two experimental groups. A difference in the overall distribution of different peaks was evident (Fig. 3A). A heat map was used to display the RPM signal value for each sample in both groups (Fig. 3B).
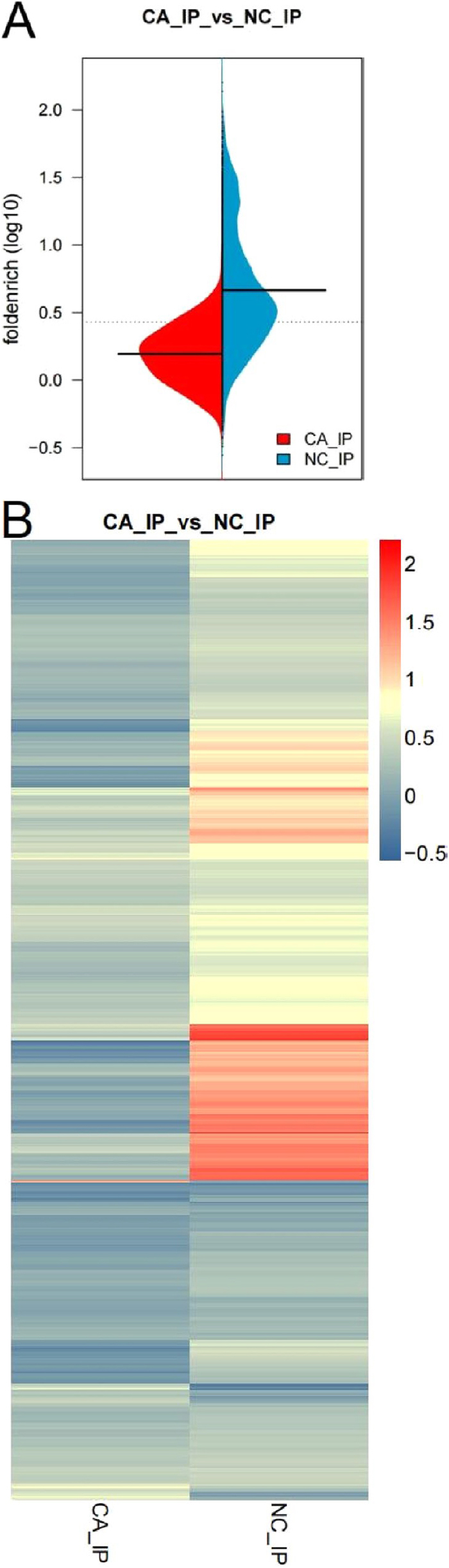
Fig. 3. Overview of differentially methylated m6A peaks modifying mRNAs. (A) The overall distribution of differential m6A peaks between the oral squamous cell carcinoma (OSCC) and adjacent normal groups by fold-enrichment. Red and blue represent the RPM distribution for the peak differences for the two groups of experiments, respectively. The black line represents the median value in each group of experiments. The dotted line represents the average value of the two groups of experiments. (B) Heat map of the signal distribution of differential m6A peaks between the OSCC and adjacent normal groups. Each row is a difference peak and each column represents each experimental group. The redder the color, the larger the RPM signal value. The bluer the color is, the smaller the RPM signal value.
To determine the role for m6A modification in OSCC, GO, and KEGG analyses were conducted on the MeRIP sequencing data. Figure 4A showed the results of a GO analysis of the genes with differential m6A peaks. The results of KEGG pathway analysis revealed that genes with upmethylation in OSCC tissues were primarily associated with the the forkhead box O (FOXO) signaling pathway (Fig. 4B). Genes with downmethylated m6A were primarily involved in the PI3K/Akt signaling pathway, pathways in cancer, the spliceosome, protein processing in the endoplasmic reticulum, and endocytosis (Fig. 4C).
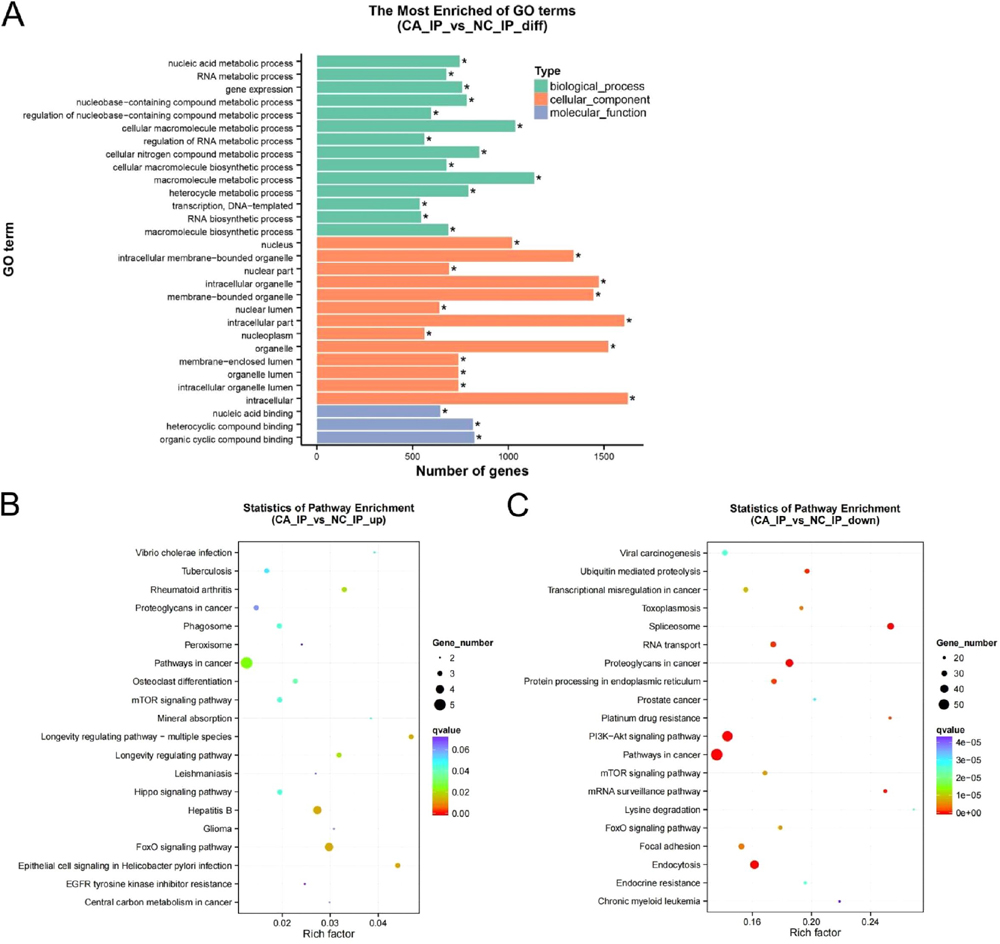
Fig. 4. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of differentially methylated mRNA. (A) The most enriched GO terms of genes with different m6A peaks. (B) The top twenty KEGG pathways for genes with upregulated m6A peaks. (C) The top twenty KEGG pathways for genes with downregulated m6A peaks.
To further explore the potential relevance between N6-methyladenosine modification and human OSCC, 24 selected genes (genes from the most prominent enriched pathways) with differential methylation peaks and 17 m6A regulators were imported into the IPA platform. As shown in Fig. 5A, METTL14, METTL3, Akt, PI3K, and SMAD2/3 form a network with WTAP. SMAD2/3 directly interacts with METTL14, METTL3, and WTAP. Akt and PI3K can indirectly inhibit WTAP, and PI3K indirectly interacts with METTL3. Figure 5B showed that YTHDF1, YTHDF3, and ALKBH5 form a network with SMAD3. YTHDF1, YTHDF3, and ALKBH5 can indirectly inhibit SMAD3.
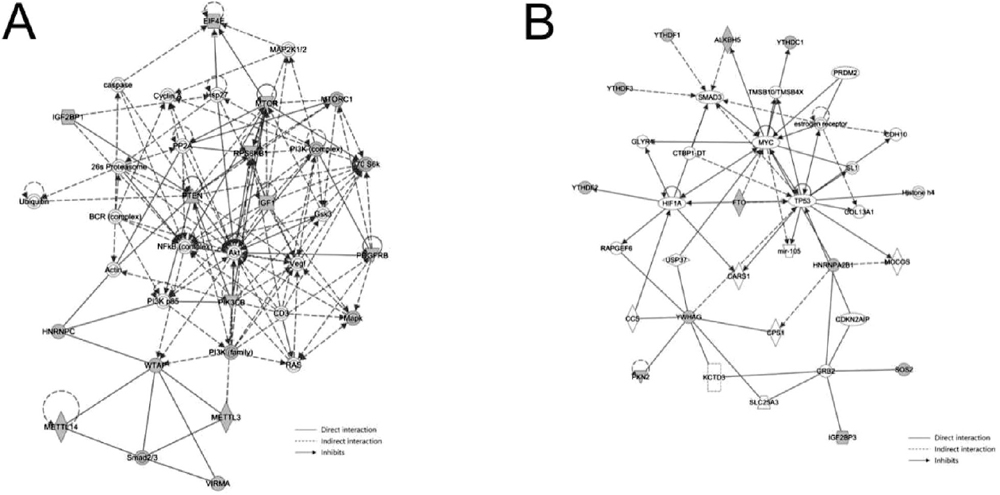
Fig. 5. Ingenuity pathway analysis of m6A methylation regulators and selected genes with differential methylation peaks. (A) METTL14, METTL3, Akt, PI3K, and SMAD2/3 form a network with WTAP. (B) YTHDF1, YTHDF3, and ALKBH5 form a network with SMAD3.
DISCUSSION
N(6)-methyladenosine modification contributes to the initiation and progression of various cancers, such as colorectal cancer (Zhang et al., 2021b), gastric cancer (Lv et al., 2020), and hepatocellular carcinoma (Wu et al., 2022). m6A modification was shown to play an important role in OSCC (Wang et al., 2021a; Xu et al., 2021), but the mRNA-wide m6A methylome of OSCC remains undefined. Using MeRIP sequencing, 2,348 dysregulated m6A peaks were identified in OSCC tissues, 85 of which showed significant upregulation and 2,263 of which showed significant downregulation. In addition, to uncover the potential biological significance of mRNAs modified by differential m6A in human OSCC, we conducted GO and KEGG pathway analyses. Moreover, IPA was used to examine the potential relevance between N6-methyladenosine modification and human OSCC. We identified several differentially methylated mRNAs associated with significant biological pathways and differentially methylated genes associated with m6A regulators.
Based on the KEGG analysis results, genes with upmethylated m6A in OSCC tissues were associated with the FOXO signaling pathway. FOXO proteins are typically associated with various cancers (Bach et al., 2018). Transcription factors encoded by the FOX gene family are associated with various cellular processes, including differentiation, DNA repair, and epigenetic modification (Lam et al., 2013). FOXM1 mRNA and protein levels in the oral cavity are increased during the initiation and progression of OSCC (Chen et al., 2014). In contrast to FOXM1, FOXO gene family members have a negative role in cellular events, particularly in promoting tumorigenesis (Jackson et al., 2010). The increased expression of FOXO3a reduces the carcinogenicity of oral cancer (Chi, 2015). In the present study, genes primarily enriched in the FOXO pathway include TGFB1, PTEN, and SOD2. TGF-B can increase the production of regulatory T-cells and inhibit the function of cytotoxic T-lymphocytes, which creates an immunosuppressive tumor microenvironment (Kondo et al., 2021). Superoxide dismutase (SOD) reduces the proliferation of cancer cells by weakening the enzymatic activity and modifying the detoxification of reactive oxygen species (Forsberg et al., 2001). PTEN is the most common mutant protein in malignant tumors and plays a negative regulatory role (Chalhoub and Baker, 2009). G protein signaling regulator 12 (RGS12) inhibits oral cancer by upregulating the phosphorylation and sulfonation of PTEN (Fu et al., 2021). Based on the present study, m6A modification may promote or inhibit OSCC tumorigenesis by attenuating the FOXO signaling pathway.
Genes that exhibit downmethylated m6A were primarily involved in PI3K/Akt signaling, the spliceosome, protein processing in the endoplasmic reticulum, and endocytosis. PI3K/Akt signaling pathway is highly correlated with the initiation and progression of various tumor types (Simpson et al., 2015; Mayer and Arteaga, 2016). The PI3K/Akt pathway plays an important role in different types of immune cells and is closely associated with an immunosuppressive tumor microenvironment. Class IA PI3Ks participate in the development and maturation of B cells as vital mediators of antigen receptor signaling (Fruman et al., 1999). With respect to the T cell compartment, PI3Ks induce the expression of cytotoxic effector molecules, such as perforin, IFNγ, and granzymes (Macintyre et al., 2011). PI3Ks also have an important role in the cytotoxicity of NK cells by inhibiting the cellular component of the innate immune response to interfere with the movement of granzyme B and perforin to target cells (Jiang et al., 2000). PI3K/Akt pathway activation sustains the secretion of suppressive cytokines, such as IL-35, TGF-b, and IL-35 to inhibit the function of effector T-cells (Soond et al., 2012). Agrin increases the expression and secretion of IL-6 through the PI3K/AKT pathway, thus promoting the development of nonsmall cell lung cancer (NSCLC) and regulating the infiltration of Tregs (Han et al., 2021). C–C Motif Chemokine Ligand 5 (CCL5) promotes an immunosuppressive tumor microenvironment by activating the PI3K/AKT pathway in clear cell renal cell carcinoma (ccRCC) cells (Xu et al., 2022). Considering that the PI3K/AKT pathway is often activated in all kinds of tumors, many studies have attempted to use PI3K/AKT inhibitors to enhance the therapeutic effect on tumors. HER3 is an upstream regulatory factor that blocks the PI3K signaling pathway. A HER3 blocking antibody plays a powerful antitumor role by inhibiting the HER3–PI3K–Akt–mTOR carcinogenic signaling, reducing the immunosuppressive microenvironment, and maintaining T cell activity (Wang et al., 2021b). Another study showed that protein histidine phosphatase (LHPP) promotes apoptosis in OSCC cells by reducing the transcriptional activities of p-PI3K and p-Akt (Liu et al., 2022). In the present study, several genes (IGF1, CREB1, and ITGB1) associated with PI3K/Akt signaling deserve attention and their relationship with PI3K/Akt signaling is shown in Supplementary Figure S2. Insulin-like growth factor I (IGF1) exerts protumorigenic effects by stimulating the proliferation and invasion of SCC-4 cells (Ferreira Mendes et al., 2020). CREB1 may be targeted by miR-654-3p to repress PSEN1 expression, which increases apoptosis in sinonasal squamous cell carcinoma (SNSCC) cells (Cui et al., 2021). Zinc finger protein 750 (ZNF750) reduces the migration of OSCC by inhibiting the cell adhesion molecule ITGB1 (Pan et al., 2018). Based on our results, regulating m6A modifications of the PI3K/Akt signaling pathway may represent an alternative therapy for OSCC.
The IPA results indicated that METTL14, METTL3, Akt, PI3K, and SMAD2/3 form a network with WTAP. Previously, it was reported that deoxycholic acid (DCA) reduces the expression of mir-92b-3p by promoting the separation of METTL3 from the METTL3–METTL14–WTAP complex, thereby increasing the protein levels of phosphatase and tensin homologues and subsequently inactivating the PI3K/AKT signaling pathway (Lin et al., 2020). SMAD2/3 can promote the formation of the METTL3–METTL14–WTAP complex with a subset of transcripts, thus disrupting the stability of specific SMAD2/3 transcriptional targets and promoting their rapid downregulation during differentiation (Bertero et al., 2018). Besides, we found that YTHDF1, YTHDF3, and ALKBH5 form a network with SMAD3. ALKBH5 overexpression weakens the stabilization of TGFβR2 and SMAD3 mRNA mediated by YTHDF1/3 and eliminates YTHDF2-mediated SMAD6 mRNA degradation, thereby inhibiting EMT induced by TGF-β and invasion of NSCLC cells. (Sun et al., 2022). Using IPA, we identified several genes with differentially methylated peaks associated with m6A regulators directly or indirectly; however, the specific mechanisms involved in their connections remain unclear. Further studies are needed to clarify the specific correlations between genes with differential m6A peaks and m6A regulators to provide new insights for exploring the pathogenesis and alternative therapies of OSCC.
Previous studies indicated that a potential strategy to target tumors is to regulate m6A methylase (METTL3, METTL14) or m6A demethylase (ALKBH5, FTO) (Zhang et al., 2021a; Guimarães-Teixeira et al., 2022; Sun et al., 2022; Yan et al., 2022). As high-throughput sequencing detection technologies are developed and more efficient computational prediction models of m6A modification are continuously improved, the potential mechanisms of N6-methyladenosine modifications in tumorigenesis and tumor progression will be elucidated. We analyzed m6A modifications of mRNAs in OSCC using MeRIP-seq. Based on our results, the direct regulation of N6-methyladenosine modifications represents an alternative therapy for OSCC.
CONCLUSIONS
In the present study, we revealed that m6A modifications play a significant role in the initiation and progression of OSCC. Our findings provide insight into OSCC epigenetic etiology and alternative therapies based on mRNA m6A methylation.
MATERIALS AND METHODS
Patients and samples
All patients signed an informed consent before tissue samples were obtained. Immediately after surgery, OSCC and adjacent normal tissues were collected and separated into centrifuge tubes. All tissue samples were stored at −80 ℃. The present study was approved by the Ethics Committee of Stomatological Hospital of Chongqing Medical University.
Library preparation
Input materials
TRIzol reagent (Invitrogen) was used to extract the total RNA from each sample. An Agilent 2100 bioanalyzer (Agilent) was used to detect the integrity of the isolated RNAs and a simpliNano spectrophotometer (GE Healthcare) was used to detect the concentration of the isolated RNAs. A Dynabeads™ mRNA Purification Kit was used to separate mRNA from extracted total RNA. Then, extracted mRNA was divided into 100-nucleotide-long fragments. Anti--m6A polyclonal antibody (Synaptic Systems) was used to enrich for m6A-methylated mRNA fragments. Next, for library construction, immunoprecipitated mRNAs were used with the NEBNext ultra RNA library prepare kit (Illumina). Based on the basis of the standard protocols (Meng et al., 2014), the HiSseq platform was used to sequence the library preparations with a paired-end read length of 150 bp.
Data analysis
Quality control
FastQC (v 0.19.11) was used to perform basic quality control on the raw data (Chen et al., 2018). Reads containing adapters or ploy-N and reads with low-quality reads were removed to obtain clean data. Meanwhile, we calculated the clean data for Q20, Q30, and GC content was calculated. The next analyses were all based on the high-quality clean data.
Reads mapping to the reference genome
The annotation documents for the gene model and reference genome were downloaded directly from the genome website. BWA (version 0.7.12) was used to build the index for the reference genome and BWA mem (version 0.7.12) was used to align the clean reads to the reference genome.
Peak calling
The m6A peaks in each group were identified with using the EexomePeak R package (v 2.16.0) and the corresponding input samples were used as controls. The q-value enrichment threshold for all data sets was 0.05. In addition, m6A motifs were identified in the mRNA regions where m6A methylation occurred by using HOMER software (version 4.9.1).
Peak annotation and different peak analysis
The genes in which the m6A peaks were located in their exons were considered to be m6A peak- related genes. And the distributions of m6A peaks in different functional regions (e.g., such as 5’UTR, CDS, 3’UTR) were performed. The ExomePeak R package (v 2.16.0) was used to perform different m6A peak calling with a fold-_change >more than 1 and a P-value <less than 0.05. Genes with differential m6A peaks (upmethylated m6A peaks or downmethylated m6A peaks) were identified using the same method and as GO and KEGG functional enrichment analyses. The GOseq R package was used to implement GO analysis, in which gene length bias was corrected and GO terms with a corrected P-value <0.05 were considered markedly enriched for genes with differential m6A peaks (Young et al., 2010). KEGG is a database that provides insight into high-level functions and utilities of biological systems, including cells, organisms, and the ecosystem, based on molecular data (http://www.genome.jp/kegg/) (Kanehisa and Goto, 2000). KOBAS software (version 3.0) was used to test the statistical enrichment of peak-related genes in the KEGG pathways.
Ingenuity pathway analysis (IPA)
Twenty-four genes exhibiting differential methylation peaks were selected from the most prominent enriched pathways and 17 m6A regulators were imported into the IPA platform to examine their potential connections.
DECLARATIONS
Funding: This study was financed by the National Natural Science Foundations of China (No. 81870775, 81500855).
Competing interests: The authors declare that they have no competing interests.
Data availability statement: The datasets generated in this study are available from the corresponding author upon reasonable request.
Author contributions: XJ designed this project. SL, YL, and HL collected and stored OSCC and adjacent normal tissues. XZ, YL, and Li Y contributed to the data analysis. YL wrote the first draft of the study. YZ, FL, and Lu Y provided valuable suggestions for the article. XJ and HL revised the manuscript. The final submitted paper was approved by all authors.
REFERENCES
- Bach, D. H., Long, N. P., Luu, T.-T.-T., Anh, N. H., Kwon, S. W., and Lee, S. K. (2018) The dominant role of forkhead box proteins in cancer. Int. J. Mol. Sci. 19, 3279.
- Bertero, A., Brown, S., Madrigal, P., Osnato, A., Ortmann, D., Yiangou, L., Kadiwala, J., Hubner, N. C., de Los Mozos, I. R., Sadée, C., et al. (2018) The SMAD2/3 interactome reveals that TGFβ controls m6A mRNA methylation in pluripotency. Nature 555, 256–259.
- Chalhoub, N., and Baker, S. J. (2009) PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 4, 127–150.
- Chen, P.-M., Cheng, Y.-W., Wang, Y.-C., Wu, T.-C., Chen, C.-Y., and Lee, H. (2014) Up-regulation of FOXM1 by E6 oncoprotein through the MZF1/NKX2-1 axis is required for human papillomavirus-associated tumorigenesis. Neoplasia 16, 961–971.
- Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018) fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890.
- Cheng, X., Li, M., Rao, X., Zhang, W., Li, X., Wang, L., and Huang, G. (2019) KIAA1429 regulates the migration and invasion of hepatocellular carcinoma by altering m6A modification of ID2 mRNA. Onco Targets Ther. 12, 3421–3428.
- Chi, H. (2015) miR-194 regulated AGK and inhibited cell proliferation of oral squamous cell carcinoma by reducing PI3K-Akt-FoxO3a signaling. Biomed. Pharmacother. 71, 53–57.
- Cristaldi, M., Mauceri, R., Di Fede, O., Giuliana, G., Campisi, G., and Panzarella, V. (2019) Salivary biomarkers for oral squamous cell carcinoma diagnosis and follow-up: current status and perspectives. Front. Physiol. 10, 1476.
- Cui, X., Yang, Y., and Yan, A. (2021) MiR-654-3p constrains proliferation, invasion, and migration of sinonasal squamous cell carcinoma via CREB1/PSEN1 regulatory axis. Front. Genet. 12, 799933.
- Dave, K., Ali, A., and Magalhaes, M. (2020) Increased expression of PD-1 and PD-L1 in oral lesions progressing to oral squamous cell carcinoma: a pilot study. Sci. Rep. 10, 9705.
- Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg, S., Cesarkas, K., Jacob-Hirsch, J., Amariglio, N., Kupiec, M., et al. (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206.
- Dreyfuss, G., Kim, V. N., and Kataoka, N. (2002) Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol. 3, 195–205.
- Du, A., Li, S., Zhou, Y., Disoma, C., Liao, Y., Zhang, Y., Chen, Z., Yang, Q., Liu, P., Liu, S., et al. (2022) M6A-mediated upregulation of circMDK promotes tumorigenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. Mol. Cancer 21, 109.
- Ferreira Mendes, J. M., de Faro Valverde, L., Torres Andion Vidal, M., Paredes, B. D., Coelho, P., Allahdadi, K. J., Coletta, R. D., Souza, B. S. F., and Rocha, C. A. G. (2020) Effects of IGF-1 on proliferation, angiogenesis, tumor stem cell populations and activation of AKT and hedgehog pathways in oral squamous cell carcinoma. Int. J. Mol. Sci. 21, 6487.
- Forsberg, L., de Faire, U., and Morgenstern, R. (2001) Oxidative stress, human genetic variation, and disease. Arch. Biochem. Biophys. 389, 84–93.
- Fruman, D. A., Snapper, S. B., Yballe, C. M., Davidson, L., Yu, J. Y., Alt, F. W., and Cantley, L. C. (1999) Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85α. Science 283, 393–397.
- Fu, C., Yuan, G., Yang, S. T., Zhang, D., and Yang, S. (2021) RGS12 Represses oral cancer via the phosphorylation and SUMOylation of PTEN. J. Dent. Res. 100, 522–531.
- Guimarães-Teixeira, C., Lobo, J., Miranda-Gonçalves, V., Barros-Silva, D., Martins-Lima, C., Monteiro-Reis, S., Sequeira, J. P., Carneiro, I., Correia, M. P., Henrique, R., et al. (2022) Downregulation of m6A writer complex member METTL14 in bladder urothelial carcinoma suppresses tumor aggressiveness. Mol. Oncol. 16, 1841–1856.
- Han, L., Shi, H., Ma, S., Luo, Y., Sun, W., Li, S., Zhang, N., Jiang, X., Gao, Y., Huang, Z., et al. (2021) Agrin Promotes Non-Small Cell Lung Cancer Progression and Stimulates Regulatory T Cells via Increasing IL-6 Secretion Through PI3K/AKT Pathway. Front. Oncol. 11, 804418.
- Jackson, B. C., Carpenter, C., Nebert, D. W., and Vasiliou, V. (2010) Update of human and mouse forkhead box (FOX) gene families. Hum. Genomics 4, 345–352.
- Jiang, K., Zhong, B., Gilvary, D. L., Corliss, B. C., Hong-Geller, E., Wei, S., and Djeu, J. Y. (2000) Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat. Immunol. 1, 419–425.
- Kanehisa, M., and Goto, S. (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30.
- Kondo, Y., Suzuki, S., Takahara, T., Ono, S., Goto, M., Miyabe, S., Sugita, Y., Ogawa, T., Ito, H., Satou, A., et al. (2021) Improving function of cytotoxic T-lymphocytes by transforming growth factor-β inhibitor in oral squamous cell carcinoma. Cancer Sci. 112, 4037–4049.
- Lam, E. W., Brosens, J. J., Gomes, A. R., and Koo, C.-Y. (2013) Forkhead box proteins: tuning forks for transcriptional harmony. Nat. Rev. Cancer 13, 482–495.
- Lee, W.-H., Chen, H.-M., Yang, S.-F., Liang, C., Peng, C.-Y., Lin, F.-M., Tsai, L.-L., Wu, B.-C., Hsin, C.-H., Chuang, C.-Y., et al. (2017) Bacterial alterations in salivary microbiota and their association in oral cancer. Sci. Rep. 7, 16540.
- Liao, L., He, Y., Li, S.-J., Zhang, G.-G., Yu, W., Yang, J., Huang, Z.-J., Zheng, C.-C., He, Q.-Y., Li, Y., et al. (2022) Anti-HIV drug elvitegravir suppresses cancer metastasis via increased proteasomal degradation of m6A methyltransferase METTL3. Cancer Res. 82, 2444–2457.
- Lin, R., Zhan, M., Yang, L., Wang, H., Shen, H., Huang, S., Huang, X., Xu, S., Zhang, Z., Li, W., et al. (2020) Deoxycholic acid modulates the progression of gallbladder cancer through N6-methyladenosine-dependent microRNA maturation. Oncogene 39, 4983–5000.
- Liu, S., Gao, W., Lu, Y., Zhou, Q., Su, R., Hasegawa, T., Du, J., and Li, M. (2022) As a novel tumor suppressor, LHPP promotes apoptosis by inhibiting the PI3K/AKT signaling pathway in oral squamous cell carcinoma. Int. J. Biol. Sci. 18, 491–506.
- Lv, Z., Sun, L., Xu, Q., Xing, C., and Yuan, Y. (2020) Joint analysis of lncRNA m6A methylome and lncRNA/mRNA expression profiles in gastric cancer. Cancer Cell Int. 20, 464.
- Macintyre, A. N., Finlay, D., Preston, G., Sinclair, L. V., Waugh, C. M., Tamas, P., Feijoo, C., Okkenhaug, K., and Cantrell, D. A. (2011) Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity 34, 224–236.
- Mascitti, M., Orsini, G., Tosco, V., Monterubbianesi, R., Balercia, A., Putignano, A., Procaccini, M., and Santarelli, A. (2018) An overview on current non-invasive diagnostic devices in oral oncology. Front Physiol. 9, 1510.
- Mayer, I. A., and Arteaga, C. L. (2016) The PI3K/AKT pathway as a target for cancer treatment. Annu. Rev. Med. 67, 11–28.
- Meng, J., Lu, Z., Liu, H., Zhang, L., Zhang, S., Chen, Y., Rao, M. K., and Huang, Y. (2014) A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/Bioconductor package. Methods 69, 274–281.
- Meyer, K. D., Saletore, Y., Zumbo, P., Elemento, O., Mason, C. E., and Jaffrey, S. R. (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 149, 1635–1646.
- Pan, L., Yang, H., Xu, C., Chen, S., Meng, Z., Li, K., and Chen, H. (2018) ZNF750 inhibited the malignant progression of oral squamous cell carcinoma by regulating tumor vascular microenvironment. Biomed. Pharmacother. 105, 566–572.
- Reyes-Gibby, C. C., Anderson, K. O., Merriman, K. W., Todd, K. H., Shete, S. S., and Hanna, E. Y. (2014) Survival patterns in squamous cell carcinoma of the head and neck: pain as an independent prognostic factor for survival. J. Pain 15, 1015–1022.
- Simpson, D. R., Mell, L. K., and Cohen, E. E. W. (2015) Targeting the PI3K/AKT/mTOR pathway in squamous cell carcinoma of the head and neck. Oral Oncol. 51, 291–298.
- Soond, D. R., Slack, E. C. M., Garden, O. A., Patton, D. T., and Okkenhaug, K. (2012) Does the PI3K pathway promote or antagonize regulatory T cell development and function? Front. Immunol. 3, 244.
- Sun, T., Wu, R., and Ming, L. (2019) The role of m6A RNA methylation in cancer. Biomed. Pharmacother. 112, 108613.
- Sun, Z., Su, Z., Zhou, Z., Wang, S., Wang, Z., Tong, X., Li, C., Wang, Y., Chen, X., Lei, Z., et al. (2022) RNA demethylase ALKBH5 inhibits TGF-β-induced EMT by regulating TGF-β/SMAD signaling in non-small cell lung cancer. FASEB J. 36, e22283.
- Tsukiyama, S., Hasan, M. M., Deng, H.-W., and Kurata, H. (2022) BERT6mA: prediction of DNA N6-methyladenine site using deep learning-based approaches. Brief. Bioinform. 23, bbac053.
- Tsukiyama, S., Hasan, M. M., and Kurata, H. (2023) CNN6mA: interpretable neural network model based on position-specific CNN and cross-interactive network for 6mA site prediction. Comput. Struct. Biotechnol. J. 21, 644–654.
- Wang, F., Zhu, Y., Cai, H., Liang, J., Wang, W., Liao, Y., Zhang, Y., Wang, C., and Hou, J. (2021a) N6-methyladenosine methyltransferase METTL14-mediated autophagy in malignant development of oral squamous cell carcinoma. Front. Oncol. 11, 738406.
- Wang, Z., Goto, Y., Allevato, M. M., Wu, V. H., Saddawi-Konefka, R., Gilardi, M., Alvarado, D., Yung, B. S., O’Farrell, A., Molinolo, A. A., et al. (2021b) Disruption of the HER3-PI3K-mTOR oncogenic signaling axis and PD-1 blockade as a multimodal precision immunotherapy in head and neck cancer. Nat. Commun. 12, 2383.
- Wu, S., Liu, S., Cao, Y., Chao, G., Wang, P., and Pan, H. (2022) Downregulation of ZC3H13 by miR-362-3p/miR-425-5p is associated with a poor prognosis and adverse outcomes in hepatocellular carcinoma. Aging (Albany NY) 14, 2304–2319.
- Xu, L., Li, Q., Wang, Y., Wang, L., Guo, Y., Yang, R., Zhao, N., Ge, N., Wang, Y., and Guo, C. (2021) m6A methyltransferase METTL3 promotes oral squamous cell carcinoma progression through enhancement of IGF2BP2-mediated SLC7A11 mRNA stability. Am. J. Cancer Res. 11, 5282–5298.
- Xu, W., Wu, Y., Liu, W., Anwaier, A., Tian, X., Su, J., Huang, H., Wei, G., Qu, Y., Zhang, H., et al. (2022) Tumor-associated macrophage-derived chemokine CCL5 facilitates the progression and immunosuppressive tumor microenvironment of clear cell renal cell carcinoma. Int. J. Biol. Sci. 18, 4884–4900.
- Yan, R., Dai, W., Wu, R., Huang, H., and Shu, M. (2022) Therapeutic targeting m6A-guided miR-146a-5p signaling contributes to the melittin-induced selective suppression of bladder cancer. Cancer Lett. 534, 215615.
- Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010) Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14.
- Zhang, J.-Y., Du, Y., Gong, L.-P., Shao, Y.-T., Pan, L.-J., Feng, Z.-Y., Pan, Y.-H., Huang, J.-T., Wen, J.-Y., Sun, L.-P., et al. (2022) ebv-circRPMS1 promotes the progression of EBV-associated gastric carcinoma via Sam68-dependent activation of METTL3. Cancer Lett. 535, 215646.
- Zhang, L., Wan, Y., Zhang, Z., Jiang, Y., Lang, J., Cheng, W., and Zhu, L. (2021a) FTO demethylates m6A modifications in HOXB13 mRNA and promotes endometrial cancer metastasis by activating the WNT signalling pathway. RNA Biol. 18, 1265–1278.
- Zhang, Z., Wang, Q., Zhang, M., Zhang, W., Zhao, L., Yang, C., Wang, B., Jiang, K., Ye, Y., Shen, Z., et al. (2021b) Comprehensive analysis of the transcriptome-wide m6A methylome in colorectal cancer by MeRIP sequencing. Epigenetics 16, 425–435.
