ORIGINAL ARTICLES
Evolutionary Analysis of Genes for S-RNase-based Self-incompatibility Reveals S Locus Duplications in the Ancestral Rosaceae
2015 Volume 84 Issue 3 Pages 233-242

Details
2015 Volume 84 Issue 3 Pages 233-242
Flowering plants have developed a genetically determined self-incompatibility (SI) system to maintain genetic diversity within a species. The Solanaceae, the Rosaceae, and the Plantaginaceae have the S-RNase-based gametophytic SI (GSI) system, which uses S-RNase and F-box proteins as the pistil S and pollen S determinants, respectively. SI is associated with culture and breeding difficulties in rosaceous fruit trees, such as apple, pear, and stone fruit species; therefore, researchers in the pomology field have long studied the mechanism and genetics of SI in order to obtain clues to overcome these difficulties. Here, we investigated the evolutionary paths of the S-RNase genes by tracking their duplication patterns. Phylogenetic analysis and estimation of proxy ages for the establishment of S-RNase and its homologs in several rosaceous species showed that the divergence of S-RNase in the subtribe Malinae and the genus Prunus predated the gene in most recent common ancestors of Rosaceae species. Furthermore, the duplicated S-RNase-like genes were accompanied by duplicated pollen S-like F-box genes, suggesting segmental duplications of the S locus. Analysis of the expression patterns and evolutionary speeds of duplicated S-RNase-like genes in Prunus suggested that these genes have lost the SI recognition function, resulting in a single S locus. Furthermore, the S loci in the current Rosaceae species might have evolved independently from the duplicated S loci, which could explain the presence of genus-specific SI recognition mechanisms in the Rosaceae. The results of the present study should be valuable for the future development of artificial SI control and for self-compatible breeding in rosaceous horticultural plant species.
Self-incompatibility (SI) is one of the most important reproductive mechanisms for maintaining genetic diversity within a species (de Nettancourt, 2001). It is widely used in angiosperms, with at least 19 orders, 71 families, and 250 genera—comprising approximately 60% of angiosperm species—showing this behavior (Allen and Hiscock, 2008). Most herbaceous annual crops lost the SI characteristic to become self-compatible (SC) during domestication because SI hinders effective crop production. In contrast, many fruit tree species and cultivars still retain SI, probably because of breeding difficulties associated with their long generation time and large size. SI is associated with difficulties in effective breeding and culture practices; therefore, its mechanism and genetics have long interested researchers in the fields of agriculture and horticulture.
In most cases, SI is genetically controlled by a single polymorphic S locus that harbors pistil S determinant and pollen S determinant genes. The mechanism can be either gametophytic (GSI) or sporophytic (SSI), depending on the genetic control of the pollen SI phenotype (Tao and Iezzoni, 2010). The Solanaceae, the Rosaceae, and the Plantaginaceae recruit the secreted cytotoxic ribonuclease (S-RNase) as pistil S determinant for their SI system, which is referred to as the S-RNase-based GSI (McClure, 2009). The pollen S determinant(s) encodes an F-box protein called SLF (S-locus F-box) in the Solanaceae and the Plantaginaceae, SFBB (S-locus F-box brothers) in the subtribe Malinae (Rosaceae), and SFB (S haplotype-specific F-box protein) in Prunus (Rosaceae) (Meng et al., 2011; Sassa et al., 2010; Tao and Iezzoni, 2010; Ushijima et al., 2003). The most recent common ancestor (MRCA) of these plant families is the root of about 75% of all dicots; therefore, S-RNase-based GSI is considered to be an ancestral state of the majority of dicots, which is supposed to have been present approximately 120 million years ago (Igic and Kohn, 2001; Steinbachs and Holsinger, 2002; Vieira et al., 2008). This would also imply that loss of the S-RNase-based GSI system and gain of another SI system could have occurred frequently during lineage speciation (Sherman-Broyles and Nasrallah, 2008).
Despite the single origin of S-RNase-based GSI and the commonality of specificity determinant genes, several lines of evidence indicate that the SI recognition mechanism in Prunus in the Rosaceae might differ from those in the other species (Hauck et al., 2006; Tao and Iezzoni, 2010; Ushijima et al., 2004). It has been proposed that the function of multiple (or a single) pollen S F-box genes at the S locus are responsible for detoxification of all but the self S-RNase in the Solanaceae, the Plantaginaceae, and the subtribe Malinae in the Rosaceae (Kakui et al., 2011; Kubo et al., 2010; Minamikawa et al., 2010; Sassa et al., 2007; Zhou et al., 2003), while a single pollen S determinant (SFB) is responsible for the cytotoxicity of the self S-RNase in Prunus (Tao and Iezzoni, 2010). This hypothesis is supported by the different outcome of hetero-diallelic pollen production and pollen S mutation in Prunus compared with the outcome in the Solanaceae, the Plantaginaceae, and the subtribe Malinae (Hauck et al., 2006; Ushijima et al., 2004). The presence of a hypothetical general inhibitor that detoxifies all S-RNase is proposed in Prunus to explain the Prunus-specific SI recognition (Tao and Iezzoni, 2010).
Gene duplication has been reported to have played an important role in the evolution of plants (Flagel and Wendel, 2009). A prevailing theory predicts gene loss (pseudogenization) or functional diversification (sub- or neofunctionalization) as the main fates for one of the two copies of duplicated genes (Blanc and Wolfe, 2004; Cusack and Wolfe, 2007; Moore and Purugganan, 2005). Of the two functional diversification mechanisms, subfunctionalization, in which each duplicate gene develops a distinct expression pattern as models of cis-evolution (Carroll, 2008), seems to be the most common phenomenon and takes place soon after gene duplication (Papp et al., 2003). In contrast, neofunctionalization is the result of diversification over the long term (Rastogi and Liberles, 2005).
Previous studies have demonstrated that S-RNase-based GSI is monophyletic and evolved in eudicots before the divergence of asterids and rosids (Igic and Kohn, 2001; Steinbachs and Holsinger, 2002; Vieira et al., 2008). Several S-RNase-like genes have been identified from certain plant species with S-RNase-based GSI. The non-S-RNases, showing homology to S-RNase, have been reported in Petunia inflata (Lee et al., 1992), Nicotiana alata (Kuroda et al., 1994), and Prunus avium (Yamane et al., 2003). The non-S-RNases found in the Solanaceae are assumed to have arisen from the duplication of the S locus, but are not associated with self-incompatibility (Golz et al., 1998; Igic and Kohn, 2001; Lee et al., 1992). Although non-S-RNase in Prunus was reported to be a possible candidate for an ancestral form of S-RNase in Prunus (Yamane et al., 2003), its physiological functions are yet to be clarified. Detailed evolutionary analysis of S-RNase and pollen S F-box genes would be useful to understand the origin and evolutionary paths of S-RNase-based GSI in the Rosaceae, and provide an insight into the molecular basis of S-RNase-based GSI in rosaceous species.
Recent advances in genome sequencing have enabled analysis of angiosperm genome evolution using genome-wide comparative analysis. According to the Phytozome database v9.1 (http://www.phytozome.net/), 41 sequenced and annotated plant genomes, which have been clustered into 20 evolutionarily significant nodes, are available. In this study, we investigated the evolutionary paths of S-RNase and its homologs, mainly by tracking their duplication patterns in genomes. Our results indicates that S-RNase and its homologs were duplicated in the ancestral genome of Rosaceae, and the S loci in the current Rosaceae species have evolved independently from the duplicated S loci. The results of this study on rosaceous S locus evolution could explain the distinct recognition mechanisms present in the Rosaceae and will be valuable for the future development of artificial SI control and SC breeding in rosaceous species.
Putative full-length sequences of 38 S-RNase-like genes were identified from the genomes of seven angiosperms and outgroup species, Selaginella and Physcomitrella, using BLASTp in Phytozome (version 9.1, http://www.phytozome.net/). We selected genes that showed significant homology (e−19 cut-off for all angiosperms and e−10 cut-off for others) to S3-RNase (accession no. AB010306) from Prunus avium. The amino acid sequences with significant BLASTp hits were subjected to gene ontology (GO) analysis by scanning the Pfam database (Finn et al., 2014) for the presence of a ribonuclease T2 motif. Information concerning the physical locations and structure of genes was based on the genome sequence obtained from the Phytozome database.
Putative full-length sequences of pollen S F-box-like genes were searched for around the regions of the S-RNase-like genes in the Rosaceae genome using Phytozome Gbrowse. We identified F-box genes showing significant homology (e−19 cut-off) to pollen S F-box genes in asterids and the subtribe Malinae. The pollen S of Prunus, SFB, was excluded from analyses in this study because it is still unclear whether Prunus SFB or asterid SLF is more closely related to Prunus SLFLs.
Construction of evolutionary topologyAlignment analyses on amino acid sequences were conducted using MAFFT ver.7 with the L-INS-I model (Katoh and Standley, 2013). The raw alignments were subjected to manual revision using Sea View ver.4 (Gouy et al., 2010). Unnecessarily long gap sequences, which disturb the proper alignment of orthologous sequences, and genes showing apparently different structures from other S-RNase-like genes were removed. The neighbor joining (NJ) approach was applied to define evolutionary topology using Mega v5.05 (Tamura et al., 2011) with 1,000 bootstrap replications.
Estimation of the proxy age of gene divergenceWe aligned the amino acid sequences of each pair of homologs using MAFFT with the L-INS-I model, and converted the amino acid alignments to nucleotide alignments using PAL2NAL (Suyama et al., 2006). We calculated the transversion rate at fourfold-degenerate sites (4DTv value) between the gene pairs using Microsoft Excel 2007 (Microsoft). Here, the fourfold-degenerate sites were the codons of amino acid residues G, A, T, P, V, R, S, and L. The 4DTv values were calculated for gene pairs retaining at least 23 fourfold-degenerate sites. For calculation of the 4DTv between specific gene groups, we averaged the 4DTv values from all combinations of the genes between the two groups. Aligned nucleotide sequences were also analyzed to calculate the values of synonymous substitutions per synonymous site (Ks), non-synonymous substitutions per non-synonymous site (Ka), and the ratio of Ka/Ks, using DnaSP 5.1 (Librado and Rozas, 2009).
Gene expression analysisTo perform organ-specific PCR analysis for S-RNase-like genes, total RNA from leaves, calyxes, petals, filaments, pollen, styles, and ovaries was isolated from P. avium (‘Satonishiki’) and P. mume (‘Nanko’) using the cold-phenol extraction method, as described by Tao et al. (1999). Total RNA of pistil tissue was also isolated from P. persica (‘Akatsuki’) and P. salicina (‘Sordum’). cDNA was synthesized from 100 ng of total RNA using the ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo, Osaka, Japan), according to the manufacturer’s protocol. Gene-specific primers for each S-RNase-like gene were designed using online software Primer3 (http://frodo.wi.mit.edu/primer3/): 5'-GTTGCCCAAGGAAAAGACAA-3' and 5'-GTCGCGTTTGTTGGAAAGAT-3' for ppa011133m (non-S-RNase1); and 5'-AGTGCTCCGACGACAAGTTT-3' and 5'-ATTGCTCGCAAAGGAGAAGA-3' for ppa024151m (non-S-RNase2). Primers 5'-ACCATAACGTTGGAGGTGGA-3' and 5'-GGAGACGAAGGACAAGGTGA-3' for ubiquitin (ppa005507m) were used as a control. Reverse transcription PCR (RT-PCR) was performed in a 20-μL reaction volume including 1× ExTaq buffer (TaKaRa Bio, Shiga, Japan), 0.2 mM dNTPs, 100 nM each of the forward and reverse primers, 0.5 U ExTaq, and cDNA equivalent to the amount synthesized from 10 ng of total RNA. The PCR cycle was: initial denaturation at 94°C for 3 min; then 35 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min; followed by final elongation at 72°C for 5 min. The PCR products were electrophoresed through a 1.5% agarose gel, stained with ethidium bromide, and visualized under UV radiation.
Identification of selective pressure on the S-RNase-like genesFull-length allele sequences of S-RNase in the Solanaceae and the subtribe Malinae, and S-RNase, non-S-RNase1, and non-S-RNase2 in Prunus, were obtained from the NCBI and Phytozome databases. Gene sequence orthologous to ppa024151m in P. avium and P. salicina were obtained by direct sequencing of PCR products. Primers were designed outside of the ORF region in ppa024151m, using sequences from the peach genome database (Genome Database for Rosaceae; GDR, Prunus_persica_v.1.0). Amino acid sequence alignments were constructed using MAFFT with the L-INS-I model, and amino acid alignments were converted to nucleotide alignments using PAL2NAL. Informative single-nucleotide polymorphisms (SNPs) in the alleles were analyzed by DnaSP 5.1 and used to calculate the evolutionary speed (Ka/Ks). Window-average Ka/Ks values were calculated from the start codon (ATG) in 90-bp windows with a 20-bp walking step, until the walking window reached the stop codon. To calculate amino acid variability level, normed variability indices (NVIs) for each site of the aligned alleles were calculated as described by Kheyr-Pour et al. (1990) using Microsoft Excel 2007. Window-average NVI values were calculated from the first amino acid (M) in the 11-AA window.
Comprehensive detection of the genes that showed significant homology to Prunus S-RNase with the ribonuclease T2 motif was performed by BLAST and gene ontology (GO) analyses. To obtain a wide range of phylogenetic data for S-RNase-like genes across angiosperms, we selected representative genomes according to Angiosperm Phylogeny Group III (APGIII) (The Angiosperm Phylogeny Group, 2009) as follows: Oryza sativa (monocot) as an outgroup of eudicots, Mimulus guttatus from asterids, Arabidopsis thaliana from malvids, and Populus trichocarpa and three genomes of the Rosaceae (Prunus persica, Fragaria vesca, and Malus × domestica) for fabits. Thirty-eight genes (one gene per locus) were identified as S-RNase-like genes.
Phylogenetic analysis of S-RNase genes in the Solanaceae, the Plantaginaceae, the subtribe Malinae, and Prunus, and the 38 S-RNase-like genes found in angiosperm genomes, indicated that the representative S-RNase-like genes in angiosperms could be divided into three major classes [Angiosperm (AG) I–III], as reported previously (Igic and Kohn, 2001) (Fig. 1). We could define at least three S-RNase-like genes in the most recent common ancestor (MRCA) of the eudicots (triangles in Fig. 1), which corresponded to the divergence of the three major classes. For the AGI and AGII classes, the divergence patterns of the genes corresponded well to the lineage speciation in APGIII. For the AGIII class, which includes S-RNases of the Solanaceae, the Plantaginaceae, and the Rosaceae, along with non-S-RNase1 (Yamane et al., 2003) from Prunus, we could find only one gene in the MRCA of eudicots, supporting the single origin of the S-RNase-based GSI system. The gene divergence patterns were, however, inconsistent with the lineage speciation, especially for the Rosaceae species, in which the S-RNases of the subtribe Malinae and Prunus diverged earlier than the root of the Rosaceae species (circle in Fig. 1).

Phylogenetic tree of S-RNase-like genes and S-RNases in the angiosperm genome. Phylogenetic tree for S-RNase-like genes from seven angiosperm genomes, along with S-RNases from asterids and Rosaceae, and outgroup species, Physcomitrella patens and Selaginella moellendorffii, constructed using amino acid sequences and the neighbor joining method. The orthologous genes in monocots are shown as stars using rice (Oryza sativa) genes. The putative genes in the MRCA of eudicots and Rosaceae are indicated by triangles and circles, respectively. The prefixes “LOC”, “mgv”, “AT”, “Potri”, “ppa”, “MDP”, and “mrna” correspond to Oryza sativa, Mimulus guttatus, Arabidopsis thaliana, Populus trichocarpa, Prunus persica, Malus × domestica, and Fragaria vesca, respectively.
The detailed phylogenetic tree constructed to focus on the AGIII class resulted in almost the same topology, with significant statistical support (Fig. 2), compared with that obtained using all S-RNase-like gene sequences (Fig. 1). Importantly, we could find only a single gene in the MRCA of Rosaceae for the non-S-RNase2 clade, which included genes from three Rosaceae species (Fragaria, Prunus, and Malus) (circled in Fig. 2). Here, we reconfirmed that the divergence of the S-RNases of the subtribe Malinae and Prunus predated the divergence of the Rosaceae species.

Phylogenetic tree, genetic distance and gene structure of S-RNase-like genes and S-RNases in Angiosperm class III. Phylogenetic tree for S-RNase-like genes in Angiosperm class III (Fig. 1), along with S-RNases from asterids and Rosaceae, and non-S-RNase1 and non-S-RNase2 genes from Prunus, constructed using amino acid sequences and the neighbor joining method. The putative genes in the MRCA of Rosaceae are shown in a circle. Intron presence/absence data for each clade are shown as a schematic diagram.
We calculated the genetic distance of each gene clade in the AGIII class (Fig. 2). The mean 4DTv value between the S-RNases from Prunus and the subtribe Malinae (0.39 ± 0.03) was significantly higher than that between the non-S-RNase2 genes from Prunus and the subtribe Malinae (0.28 ± 0.03). Furthermore, the mean 4DTv value between mrna00224.1-v1.0-hybrid and other genes in the non-S-RNase2 clade (0.42 ± 0.01) was higher than that between the non-S-RNase2 genes from Prunus and the subtribe Malinae, while no significant difference was observed between the mean 4DTv value between mrna00224.1-v1.0-hybrid and other genes in the non-S-RNase2 clade and that between the S-RNases from Prunus and the subtribe Malinae. The mean Ks value between the S-RNase genes from Prunus and the subtribe Malinae (0.78 ± 0.004) was significantly higher than that between the non-S-RNase2 genes from Prunus and the subtribe Malinae (0.61 ± 0.015) and that between mrna00224.1-v1.0-hybrid and other genes in the non-S-RNase2 clade (0.70 ± 0.013) (Table 1). Collectively, these results supported the topology of the phylogenetic tree (Fig. 2), and indicated that the current S-RNases of the subtribe Malinae and Prunus may have been generated by a gene duplication event that occurred before the establishment of the Fragaria, Prunus, and Malinae.

Ka/Ks values for S-RNase and S-RNase-like genes in Rosaceae.
It was reported that S-RNases in asterids and the subtribe Malinae have one intron, whereas Prunus S-RNase has an additional intron located in the upstream region of the common intron present in all S-RNases (Igic and Kohn, 2001). Prunus non-S-RNase1, on the other hand, has only one intron, located at the same position as the introns of asterids and the subtribe Malinae S-RNases (Yamane et al., 2003). The genes in the non-S-RNase2 clade also have only one intron at the same position as the common introns of all S-RNases, indicating that non-S-RNases have maintained the original structure of S-RNase (Fig. 2). The additional intron found only in Prunus S-RNase may have been produced after the gene duplication that generated non-S-RNase1 and Prunus S-RNase.
Evolutionary patterns of pollen S F-box-like genesIn the surrounding regions of the S-RNase-like genes in the Rosaceae, we found F-box genes showing significant homology to pollen S F-box genes in asterids and the subtribe Malinae (Fig. 3). The phylogenetic trees constructed from pollen S F-box-like genes and their counterpart S-RNases showed almost the same topologies, with significant statistical support (Figs. 2 and 3), suggesting that the F-box/RNase segments of the S locus were duplicated. Conversely, no F-box gene was detected in the proximity of the non-S-RNase1 gene in the P. persica genome.

Phylogenetic tree and gene location of pollen S F-box-like and pollen S F-box genes in Prunus, Fragaria, and the subtribe Malinae in the Rosaceae, along with the asterid pollen S F-box gene. For pollen S F-box genes in the Solanaceae and the tribe Malinae, we included all SLFs identified in the S11 haplotype of Petunia × hybrida (Kubo et al., 2015) and all SFBBs from the S3 haplotype of Pyrus pyrifolia (Kakui et al., 2011). The phylogenetic tree was constructed using amino acid sequences and the neighbor joining method. The pollen S of Prunus, SFB, was not used to construct this tree because it is still unclear whether Prunus SFB or asterid SLF should be used as the outgroup. The putative genes in MRCA of Rosaceae are shown in a circle. Each symbol in the phylogenetic tree corresponds to those in the schematic diagram of S-RNase evolution shown in the inset figure (upper left).
To explore the possibility of change in cis-functions between duplicated S-RNase-like genes, expression analysis was conducted with non-S-RNase1 and non-S-RNase2 genes of Prunus species. Although the S-RNase-like genes were expressed mainly in the style (Fig. 4A), the expression patterns differed among species in Prunus (Fig. 4B). Expression of non-S-RNase1 (ppa011133m) was detected in the styles of P. avium, P. mume, and P. persica, but not in those of P. salicina. The non-S-RNase2 (ppa024151m) showed significant expression in the styles of P. mume and P. persica, but not in those of P. avium and P. salicina.

Expression profiling of the S-RNase-like genes in Prunus. (A) Organ-specific expression profiles of non-S-RNase1 and non-S-RNase2 in P. avium (left) and P. mume (right). RT-PCR was performed on samples from leaves (Lf), calyx (C), petal (Pe), filament (Fi), pollen (Po), style (Sty), and ovary (Ov). The expression of ubiquitin (ppa005507m) was used as a reference control. (B) Species-specific expression of S-RNase-like genes was determined by RT-PCR using cDNA prepared from the styles of P. persica, P. salicina, P. avium, and P. mume.
Selective pressure on non-S-RNase1 and non-S-RNase2 genes in Prunus, and S-RNases in asterids, in the subtribe Malinae, and in Prunus, was investigated in terms of the Ka/Ks and NVI, which were previously used to assess the amino acid variable sites of S-RNases (Ioerger et al., 1991; Kheyr-Pour et al., 1990; Ushijima et al., 1998). A sliding window analysis indicated that Ka/Ks peaks appeared in the hypervariable (HV) region (Ishimizu et al., 1998a, b; Ushijima et al., 1998) in S-RNases of asterids and Prunus, while no such indication was observed for S-RNases of the subtribe Malinae (Fig. 5A). The window-averaged plot of NVI clearly indicated that the HV region of all S-RNases showed significantly high NVI values (NVI > 0), indicating positive selection in this region (Fig. 5B). No sign of positive selection was detected in the region corresponding to the Rosaceae hypervariable (RHV) regions of duplicated S-RNase-like genes.
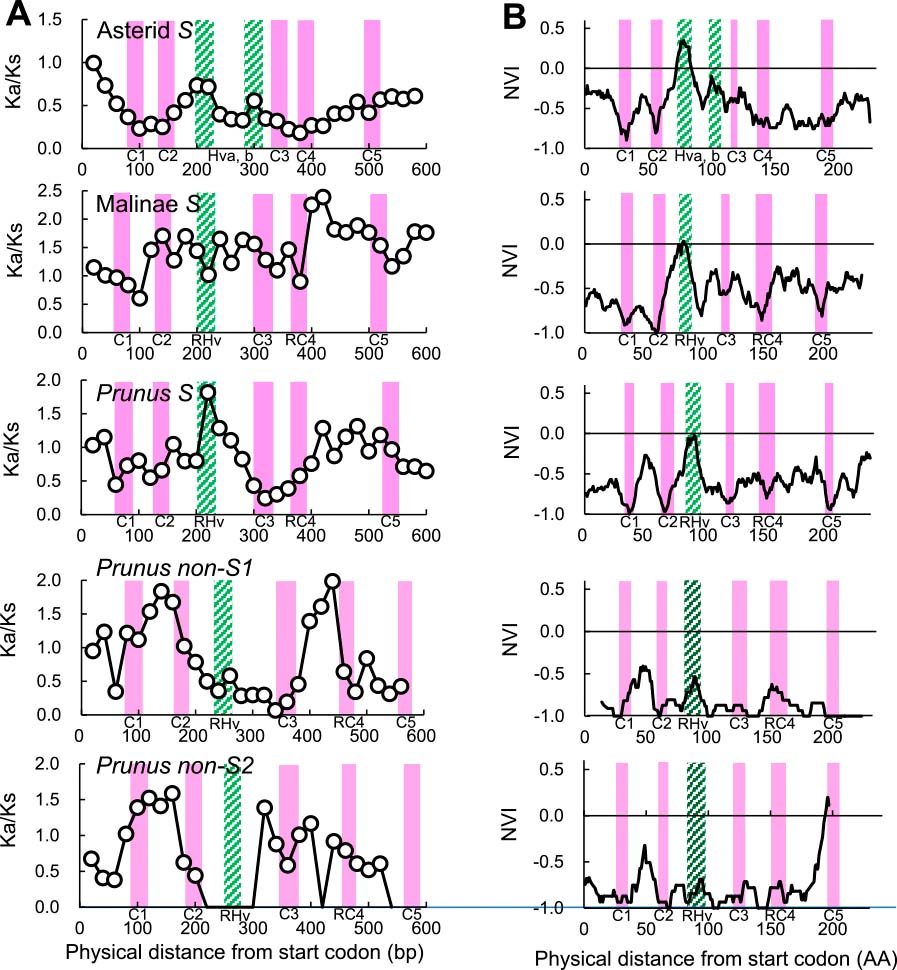
Selective pressure on S-RNase-like genes in Prunus and S-RNases in asterids, the subtribe Malinae, and Prunus. Window-average Ka/Ks and NVI values in the S-RNase gene of asterids, the subtribe Malinae, and Prunus, and non-S-RNase1 and non-S-RNase2 in Prunus. Conserved (C1–C5) and hypervariable (HV) regions are shown by closed and hatched bars, respectively. For non-S-RNase1 and non-S-RNase2, those regions were estimated by alignment with the S-RNase sequence. (A) Window-average Ka/Ks values in 90-bp sliding windows with a 20-bp walking step. (B) Window-average NVI values in 11-AA sliding windows.
Previous studies suggested that the S-RNase-based GSI system evolved once in eudicots (Igic and Kohn, 2001; Steinbachs and Holsinger, 2002; Vieira et al., 2008). Consistent with this, our results supported a single origin of the S-RNase-based GSI system in eudicots (Figs. 1 and 2). However, we identified segmental duplications of the S locus in the Rosaceae lineage. Figure 6 shows a schematic model of the duplications of S-RNase and pollen S F-box genes. The current S-RNases in asterids and Rosaceae are thought to have originated from a single original S-RNase. After the divergence of the asterid and rosid ancestors, the original S-RNase was duplicated in the ancestral early-stage genome of the Rosaceae, producing the S-RNases of the subtribe Malinae (Dupli. S-I in Fig. 6). Subsequently, another ancestral S-RNase produced by this duplication was thought to have been duplicated again to produce the ancestral S-RNases of Prunus and the non-S-RNase2 genes (Dupli. S-II). Note that we could not find genes orthologous to the S-RNases of the subtribe Malinae in Prunus and Fragaria, nor those to S-RNase of Prunus in the subtribe Malinae and Fragaria. Similar patterns of duplication and evolutionary paths have been suggested for the S locus F-box gene divergence. As observed in the evolutionary paths of S-RNases, two duplications that generated SFBB of the subtribe Malinae and Prunus SLFLs (Entani et al., 2003; Matsumoto et al., 2008; Ushijima et al., 2003) were defined by Dupli. S-I and Dupli. S-II, respectively. Within the range of the genes analyzed in this study, the origin of SFB, the pollen S F-box gene of Prunus, remains unknown.

Schematic diagram of the establishment of S-RNase and pollen S F-box genes in the Rosaceae. After the divergence of the asterid and rosid ancestor, the first S-RNase duplication occurred in the early-stage ancestral genome of Rosaceae, producing the S-RNase of the subtribe Malinae (Dupli. S-I). Subsequently, another S locus was duplicated into the ancestral S locus of Prunus and non-S-RNase2 (Dupli. S-II). A similar evolutionary pattern was found for pollen S F-box gene divergence. Two duplication events that generated SFBB of the subtribe Malinae and SLFLs of Prunus correspond to Dupli. S-I and Dupli. S-II, respectively, indicating the segmental duplication of the S locus. Prunus S-RNase lineage-specific gene duplication may have produced non-S-RNase1 of Prunus before Rosaceae species divergence (triangle). Dotted line indicates the time of the establishment of Fragaria, Prunus, and the subtribe Malinae. Putative models of the evolution of the S-RNase and pollen S F-box genes in Fragaria are shown in thin lines as suspicious branches. Within the range of the genes used in this study, the origin of the pollen S of Prunus, SFB, could not be determined.
Recent studies suggested that duplicated genes experience subfunctionalization, mainly in their cis-functions (Liu and Adams, 2010; Roulin et al., 2013). This study demonstrated that non-S-RNase1 and non-S-RNase2 genes, which are S-RNase-like genes, showed style-specific expression in some Prunus species, as did S-RNase in Prunus, while expression was not detected in styles of other Prunus species tested, indicating that cis-evolution or pseudogenization had taken place in the duplicated genes. Regarding the maintenance of the trans-functions in duplicated S-RNase-like genes, no sign of positive selection (balancing selection) on the HV region was observed for non-S-RNase1 and non-S-RNase2 genes. Given that the HV region is necessary for self/nonself recognition (Ishimizu et al., 1998a, b; Ushijima et al., 1998), the duplicated S-RNase-like genes are thought to have lost the self/nonself recognition function, leading to the presence a single S locus state in Prunus.
Recently, a diploid Fragaria was reported to have two independent self-incompatibility loci (S and T) controlling S-RNase-based GSI (Bošković et al., 2010). Comparative mapping analysis showed partial synteny between the T (LG6) and S (LG1) loci and Prunus LG6 on which the S locus lies (Bošković et al., 2010). In our phylogenetic analysis, five genes were identified as S-RNase-like genes in the genome of Fragaria vesca, of which mrna00224 and mrna00227 are located around the T locus on chromosome 6 and are clustered closely together with the Prunus S-RNase in the phylogenetic tree. Mrna13010, located on chromosome 1, appeared to diverge at an early stage of the ancestral genome of the Rosaceae. Therefore, it is possible that the S-RNase-based GSI system in Fragaria has evolved differently from those of the subtribe Malinae and Prunus. It should be noted, however, that Fragaria vesca is an SC species, and was reported to have no RNase activity in its style (Bošković et al., 2010). No mutation producing a premature stop codon could be observed in coding sequences of mrna00224, mrna00227, and mrna13010 within the range of our analyses, so it is possible that there are other mutations that affect RNase activity or, alternatively, these genes are not related to self-incompatibility of Fragaria.
In conclusion, the S locus and its specificity determinant genes have experienced several duplications during the evolution of the Rosaceae. Furthermore, the S-RNase-based GSI in the Rosaceae species have evolved independently from the duplicated S loci, recruiting different duplicated genes, which may explain the genus-specific SI recognition systems present in the Rosaceae. The genus-specific SI recognition mechanisms, in turn, should be considered for the future development of artificial SI controls and for SC breeding.