Abstract
The Brassica genus comprises various important species, of which three diploid species, B. rapa (A genome), B. nigra (B), and B. oleracea (C), yielded three different pair-wise amphidiploids: B. juncea (AB), B. napus (AC), and B. carinata (BC), showing the “triangle of U”. Although DNA sequences of many genes have been analyzed to reveal the relationships between A, B, and C genomes, the phylogeny of any single-copy nuclear gene has not supported the entire relationships of U’s triangle. Most nuclear genomic sequences of plants have genetically recombined between alleles in inter-specific hybrids, while we recently found that intron 19 and nucleotide tag (Ntag) sequences of the single-copy nuclear PolA1 gene, encoding RNA polymerase I’s largest subunit, had rarely recombined during the introgressive hybridizations in Aegilops speltoides. Because phylogenetic analysis including recombined sequences cannot reveal the phylogeny before the recombination occurred, only analysis of non-recombinational DNA sequences can resolve the true evolutionary route. In this study, the phylogenetic relationships of the PolA1 gene in the six Brassica species were clearly consistent with U’s triangle. In addition, two groups of B. napus were shown divergently to have originated from the amphidiploidization between B. oleracea and two progenitors of B. rapa.
Introduction
The family Brassicaceae contains about 338 genera and 3709 species (Al-Shehbaz et al., 2006). Three amphidiploid species, namely, B. juncea (2n = 36, AABB), B. napus (2n = 38, AACC), and B. carinata (2n = 34, BBCC), were derived from hybridization between diploid species, namely, B. rapa (2n = 20, AA), B. nigra (2n = 16, BB), and B. oleracea (2n = 18, CC) (Morinaga, 1934; U, 1935). U’s triangle hypothesis has been partly supported by various studies using different criteria, such as flavonoid composition (Dass and Nybom, 1967), seed protein serology (Vaughan, 1977), and isozymes (Takahata and Hinata, 1986). The hypothesis has also been confirmed in part by several molecular studies using restriction fragment length polymorphism (Song et al., 1988), random amplified polymorphic DNA (Demeke et al., 1992), and plastid DNA (Flannery et al., 2006; Palmer et al., 1983). Sequence analysis, however, of any single-copy nuclear gene has not proven the phylogenetic relationships of U’s triangle in Brassica species.
Because of the economic importance of Brassicaceae crops, extensive studies have been conducted to elucidate their chromosomal variations, cytological relationship, and inter-specific hybridization of different species (Mizushima, 1980). It has also been suggested (Prakash and Hinata, 1980) that the genome of the diploid species has evolved from an ancestral Brassica with n = 9. On the basis of its morphology (Prakash and Hinata, 1980), cultivated B. rapa can be divided into two diverse groups, one in Europe and the other in South China. Homology between B. rapa (A genome) and B. oleracea (C genome) shows that they are the descendants of the same ancestor, distant to B. nigra (B genome) (Chevre et al., 1991; Song et al., 1988).
Genomic DNA sequences of plants evolve through two different modes: 1) changes of DNA sequence by natural mutations, such as base substitution, insertion, and deletion, and 2) genetic recombination between two different allelic DNA sequences during meiosis in an inter-specific hybrid. In principle, phylogenetic algorithms have to analyze sequences derived from a common ancestral sequence, not genetically recombined sequences. The Brassica species were probably established through repeated inter-specific hybridizations (overviewed by Organisation for Economic Cooperation and Development (OECD), 2012) and are of mesopolyploid origin (Wang et al., 2011). The phylogeny of no nuclear gene has been consistent with all relationships of U’s triangle. Therefore, we consider that only the analysis of DNA sequences with no genetic recombination can resolve the evolutionary route.
PolA1 is a single-copy nuclear gene in the diploid genome of plants and encodes the largest subunit (POLA1) of RNA polymerase I complex. The PolA1 gene consists of 21 exons and spans approximately 9.0 kb in Arabidopsis thaliana, which is a model plant of the Brassicaceae family. Sequence polymorphisms of PolA1 intron 19 were found to be useful for revealing the phylogenetic relationships among species of Petunia (Zhang et al., 2008), Oryza (Takahashi et al., 2009), Triticum-Aegilops (Takahashi et al., 2010). Rai et al. (2012) showed that Aegilops speltoides is an introgressive species because it maintains the intron 19 and nucleotide tag (Ntag) sequences of the PolA1 gene from its progenitor in the genus Hordeum. These results indicated that these sequences of the PolA1 gene have rarely recombined during the successive backcrosses by Triticum-Aegilops species.
In this study, the intron 19 and Ntag sequences of the PolA1 gene were analyzed to reveal the relationship among three diploid and three tetraploid species, constituting members of the triangle of U in Brassica.
Materials and Methods
Plant material and DNA extraction
Twenty-eight accessions of Brassica comprising eight tetraploid and twenty diploid accessions were used in this study. Ten accessions of B. rapa (A genome), five accessions of B. nigra (B), and five accessions of B. oleracea (C) were analyzed (Table 1). For amphidiploid species, four accessions of B. napus (AC), two accessions of B. juncea (AB), and two accessions of B. carinata (BC) provided from the Brassica Seed Collection of Tohoku University were used.
Approximately 100 mg of young leaves were frozen in 2 ml plastic tubes with liquid nitrogen and crushed into a fine powder using MULTI-BEAD SHOCKER (Yasui Kikai Co., Osaka, Japan). Genomic DNA was extracted using the CTAB method (Doyle and Doyle, 1987).
Amplification and analysis of intron 19 sequence of PolA1 gene
The DNA fragments containing the intron 19 sequences were amplified by PCR using primers, 19int5P and 19int3P, located on exons 19 and 20 of the PolA1 gene, respectively (Fig. 1). PCR amplification was performed with ExTaq DNA polymerase (TaKaRa Co., Shiga, Japan) according to the manufacturer’s instructions. The thermocycling profile consisted of an initial denaturation step at 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 1 min, annealing at 59°C for 1 min, and extension at 72°C for 2 min in a PTC200 thermocycler (MJ Research Inc., Woburn, MA, USA). The amplified PCR products were subjected to 2% agarose gel electrophoresis and purified using a PCR purification kit (Qiagen Inc., Valencia, CA, USA).

The purified PCR products of diploid species were sequenced directly using the same primers as used for PCR amplification in an automated DNA sequencer (ABI310; Life Technologies, Carlsbad, CA, USA) with a BigDye Terminator Cycle Sequencing Kit (Life Technologies). For amphidiploid genome species, PCR products were amplified using a pair of primers, 19int5P and 19int3P. The purified PCR product was cloned into the pUC19 plasmid and five clones were sequenced using the M13 RV sequencing primer.
Analysis of Ntag sequence of PolA1 gene and RT-PCR of genome-specific cDNA fragments
As shown in Figure 1, the Ntag sequence is a protein-coding sequence delimited between highly conserved NBL (60 bp in the 19th exon) and NBR (48 bp in the 21st exon) sequences of the PolA1 gene (Nakamura, 2010; Rai et al., 2012). Total RNA was extracted from leaves by using Plant RNA Reagent (Life Technologies) followed by DNase treatment. RT-PCR was carried out using Invitrogen Superscript III Reverse Transcriptase kit (Life Technologies). The Ntag sequence (ca. 1.2 kb) was amplified using cDNA as the template and primers, 19ex5P and 21ex3P (Table 2), and determined by direct sequencing using the same primers. RT-PCR was also employed to analyze the expression of each PolA1 gene in three tetraploid species using three different genome-specific primers (RapRT5P, OleRT5P, and NigRT5P) and one common primer (ABCRT3P), located on exons 20 and 21, respectively (Fig. 1).
Phylogenetic analysis of intron 19 and Ntag sequences of PolA1 gene
PolA1 intron 19 sequences of Brassica species were aligned using Genetyx Software ver. 15.05 (Software Development Co., Tokyo, Japan). The determined Ntag sequences were aligned using CLUSTALW (Thompson et al., 1994). Phylogenetic trees of intron 19 and Ntag sequences were constructed by the UPGMA and neighbor-joining methods (Saitou and Nei, 1987) with bootstrap analysis using 1000 replicates of MEGA5 software (Tamura et al., 2011), respectively.
Results
Amplification of DNA fragment containing intron 19 of PolA1 gene
Amplified PCR products of the intron 19 sequences showed four differently sized bands in the three diploid Brassica species (Fig. 2). Accessions of B. rapa (A genome) had either the Rapa-L or the Rapa-S band. The Rapa-L band was found in ‘Tatsoi’ of var. narinosa, ‘Atsumi kabu’ of var. rapa, and two accessions of var. pekinensis, whereas the remaining accessions of vars. chinensis, parachinensis, pekinensis, and rapa showed the Rapa-S band (Table 1). Brassica nigra (B genome) had the smallest band (Nig), while B. oleracea (C genome) showed the second largest one (Ole).

Four accessions of B. napus (AC genome) showed two bands, the sizes of which were identical to either the Rapa-L or the Rapa-S band of B. rapa and the Ole band of B. oleracea, although the additional largest band was an artificial PCR product, which was probably made from two other PCR products (Fig. 2). Two accessions of B. carinata (BC genome) had two PCR products of the Nig band of B. nigra and the Ole band of B. oleracea, while two accessions of B. juncea (AB genome) contained the Rapa-S band of B. rapa and the Nig band of B. nigra.
Alignment of P119 sequence among the three diploid species
PolA1 intron 19 sequences were aligned among two accessions, ‘Tatsoi’ (Rapa-L) and ‘Yukina’ (Rapa-S), in B. rapa, one accession (Ni138) in B. nigra, and one accession (O169) in B. oleracea (Fig. 3). These four sequences were highly homologous at both ends, but differed in length due to various insertions and deletions. Interestingly, all intron 19 sequences of diploid and tetraploid species showed four particular lengths, that is, Rapa-L (242 bp), Rapa-S (183 bp), Ole (221 bp), and Nig (111 or 112 bp), as listed in Table 1.
Similarities of PolA1 intron 19 sequences in Brassica species
In order to analyze the similarity of the intron 19 sequences, a phylogenetic tree was constructed using the UPGMA option in MEGA5 software (Fig. 4). The intron 19 sequences of B. rapa were clearly classified into two groups, Rapa-L and Rapa-S. The Rapa-L, Rapa-S, and Ole groups contained 6, 10, and 11 intron 19 sequences, which shared high homology. In contrast, 8 intron 19 sequences of the Nig group were divided into three subgroups, Nig1, Nig2, and Nig3. The sequences of the Rapa-L group had closer relationships with those of the Ole group than the Rapa-S group.

Two accessions (N117, N127) of B. napus had two intron 19 sequences, which belonged to the Rapa-L and Ole groups, while the other two accessions (N131, N135) contained the two sequences of the Rapa-S and Ole groups (Fig. 4). Two accessions (J113, J473) of B. juncea had the two sequences of the Rapa-S and Nig3 groups. One accession (Ca105) of B. carinata showed the two sequences of the Ole and Nig1 groups. Only one sequence of the Ole group could be determined in the Ca112 accession.
Phylogenetic relationships among Ntag sequences in Brassica species
The neighbor-joining tree of Ntag sequences indicated that 10 accessions of B. rapa were clearly separated into two groups, Rapa-L and Rapa-S (Fig. 5). Although there were several polymorphisms among Ntag sequences within the Rapa-L and Rapa-S groups, only one non-synonymous substitution was found between Rapa-L (Ala) and Rapa-S (Thr), except for a 9-base deletion found in the Rapa-L group. The Ntag sequences of B. oleracea showed closer relationships with those of Rapa-L and Rapa-S than B. nigra. The Ntag sequence of Raphanus sativus was closely related to three Brassica species when Eutrema salsugineum was used as an outgroup.
Variations of Ntag sequences of PolA1 genes
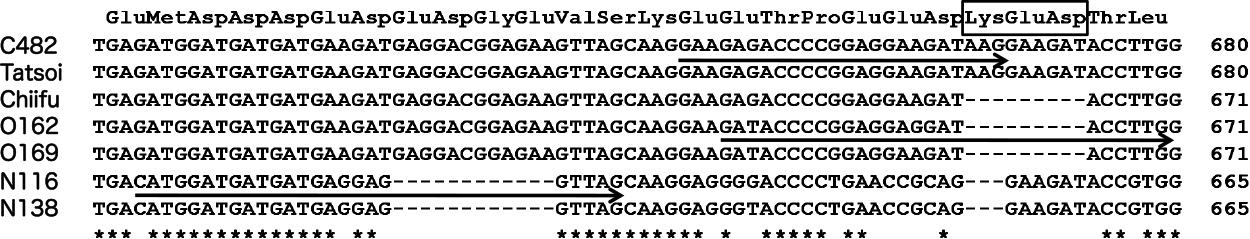
Highly polymorphic alignment among Ntag sequences (600 bp to 680 bp) of three diploid Brassica species is shown in Figure 6. The Ntag sequences of ‘C482’ (Rapa-S group) and ‘Tatsoi’ (Rapa-L group) were identical in this region. Two accessions, ‘Chiifu’ and ‘Matsushima hakusai’ (Rapa-L group), of Chinese cabbage (B. rapa var. pekinensis) had Ntag sequences homologous to that of ‘Tatsoi,’ but they shared a 9-base deletion, encoding three amino acids, Lys-Glu-Asp, which was also shared with two accessions (O162, O169) of B. oleracea. Two accessions (N116, N138) of B. nigra had two species-specific deletions (12 bases and 3 bases). On the basis of these sequence polymorphisms, three RT-PCR primers, RapRT5P, OleRT5P, and NigRT5P (Fig. 1; Table 2), were designed to amplify mRNA from the genome-specific PolA1 genes of B. rapa, B. oleracea, and B. nigra, respectively.
RT-PCR of genome-specific PolA1 mRNA
As shown in Fig. 7, three different RT-PCR products that were A-genome-specific 337 bp (B. rapa), B-genome-specific 359 bp (B. nigra), and C-genome-specific 325 bp (B. oleracea) were amplified by RT-PCR using three 5P primers (RapRT5P, OleRT5P, and NigRT5P) and a common 3P primer (BraABCRT3P). In the three amphidiploid species, two RT-PCR products were amplified in B. napus with a combination of A- and C-, in B. carinata with a combination of B- and C-, and in B. juncea with a combination of A- and B-genome-specific bands. The combinations of genome-specific RT-PCR products from the PolA1 gene in the three amphidiploid species were consistent with the relationships of U’s triangle.
Discussion
Particular length of intron 19 sequence of PolA1 gene
Whole genome sequencing analyses of B. rapa and B. oleracea indicated that three Brassica diploid species had triplicated genomes, which originated from inter-specific hybridizations among three ancestral species (Town et al., 2006; Wang et al., 2011). If the three ancestral species were involved in the origin of diploid Brassica species, two PolA1 genes had been lost during the speciation. In this study, B. oleracea and B. nigra showed particular lengths, 221 bp (Ole) and 111–112 bp (Nig), of the intron 19 sequence of the PolA1 gene, respectively, whereas B. rapa had two discrete lengths, 183 bp (Rapa-S) and 242 bp (Rapa-L), of the intron 19 sequence (Table 1). These results suggest that the intron 19 sequences had not been genetically recombined among the three ancestral PolA1 genes during the origin of the three diploid Brassica species.
Although the additional band was an artificial PCR product (Fig. 2), an accession (Ca105) of B. carinata (BC genome) had two bands of Ole (221 bp) and Nig1 (111 bp), as listed in Table 1. Two accessions (J113, J473) of B. juncea (AB genome) showed two PCR products, Rapa-S (183 bp) and Nig3 (112 bp), whereas two accessions (N117, N127) of B. napus (AC genome) had two PCR products, Rapa-L (242 bp) and Ole (221 bp), and two other accessions (N131, N135) showed two PCR products, Rapa-S (183 bp) and Ole (221 bp). This result indicates that B. napus has diphyletic origins, which was also suggested by an analysis of the S-locus gene (Okamoto et al., 2007).
The overall composition of the intron 19 sequences in the six Brassica species was clearly consistent with the entire relationships of the triangle of U (U, 1935). In addition, we found that two groups of B. napus divergently originated through the amphidiploidization between B. oleracea and two groups (Rapa-S or Rapa-L) of B. rapa. This result indicated that the intron 19 sequence of the PolA1 gene was not recombined during the amphidiploidization and is a useful DNA marker to resolve the origin of amphidiploid species, such as Brassica tetraploid species.
Variations of Ntag sequence of PolA1 gene
In contrast to the intron 19 sequences, Ntag sequences of the Rapa-L group had closer relationships to those of the Rapa-S group than the Ole group (Fig. 5). Brassica rapa complex originated from inter-specific hybridization between two closely related progenitors. Out of 4 accessions of B. rapa var. pekinensis, two accessions belonged to the Rapa-S group and the remaining two accessions, ‘Chiifu’ and ‘Matsushima hakusai’, had the Rapa-L-type Ntag sequence with a 9-base deletion, which was shared with two accessions of B. oleracea (Fig. 6). These results suggest that Chinese cabbage probably originated from hybridization between Rapa-S- and Rapa-L-type progenitors, and the Rapa-L-type progenitor might have been introgressed by B. oleracea.
RT-PCR analysis of PolA1 mRNA
On the basis of polymorphisms found in the Ntag sequences of PolA1 genes among B. rapa, B. oleracea, and B. nigra (Fig. 6), RT-PCR was conducted to amplify the genome-specific cDNA fragment from each PolA1 gene. Single RT-PCR product of A-genome-specific (337 bp), B-genome-specific (359 bp), and C-genome-specific (325 bp) were amplified in B. rapa, B. nigra, and B. oleracea, respectively (Fig. 7). This result also suggests that the active PolA1 gene is a single copy in each diploid species.
Two RT-PCR products that were A- and C-genome-specific, B- and C-genome-specific, and A- and B-genome-specific were detected in B. napus, B. carinata, and B. juncea, respectively. This single gel picture clearly supported all relationships of U’s triangle. These findings showed that two genome-specific PolA1 genes were expressed in each of the three amphidiploid species.
Brassica rapa has substantial polymorphism due to its diverse background and wide geographical distribution. Nishi (1980) proposed that primitive-type B. rapa originated in Europe, migrated to Asia, and then differentiated from its primitive type. It was observed that B. rapa exhibited more variability in both nuclear and mitochondrial DNA than B. oleracea. In this study, as two types of PolA1 genes were found in B. rapa, this species was suggested to have originated from hybridizations between two progenitors, Rapa-L and Rapa-S groups. This probably explains why B. rapa shows wide variations.
Although the Brassica species have evolved through inter-specific hybridizations and chromosome doubling, the intron 19 and Ntag sequences of the PolA1 gene have rarely genetically recombined, the reason for which remains to be resolved. The nucleotide sequence of a single-copy nuclear gene is useful as a constructive marker to evaluate phylogenetics. Single-copy genes, however, usually encode housekeeping proteins that are highly conservative. Thus, housekeeping genes that have genetically recombined by inter-specific hybridization can still encode the functional protein. Therefore, most single-copy genes except the PolA1 gene have probably recombined during the evolution of the Brassica species.
In this study, the phylogeny of the PolA1 gene was consistent with all relationships of the triangle of U (Fig. 8). In addition, it suggests that B. rapa originated from the hybridization between two progenitor species, which were independently involved in the origin of B. napus. Combined analyses of the PolA1 gene and entire genome sequences should contribute to resolution of the introgressive speciation, which is probably a more common mechanism of plant evolution than geographical speciation.
Acknowledgements
We are grateful to Professor Takeshi Nishio, Tohoku University Brassica Seed Collection, for providing seeds of Brassica species.
Literature Cited
- Al-Shehbaz, I. A., M. A. Beilstein and E. A. Kellogg. 2006. Systematics and phylogeny of the Brassicaceae (Cruciferae): an overview. Plant Syst. Evol. 259: 89–120.
- Chevre, A. M., P. This, F. Eber, M. Deschamps, M. Renard, M. Delseny and C. F. Quiros. 1991. Characterization of disomic addition lines Brassica napus-Brassica nigra by isozyme, fatty acid, and RFLP markers. Theor. Appl. Genet. 81: 43–49.
- Dass, H. and N. Nybom. 1967. The relationships between Brassica nigra, B. campestris, B. oleracea and their amphidiploid hybrids studied by means of numerical chemotaxonomy. Can. J. Genet. Cytol. 9: 880–890.
- Demeke, T., R. P. Adams and R. Chibbar. 1992. Potential taxonomic use of random amplified polymorphic DNA (RAPD): a case study in Brassica. Theor. Appl. Genet. 84: 990–994.
- Doyle, J. J. and J. L. Doyle. 1987. A rapid DNA isolation procedure for small quantities of fresh Leaf tissue. Phytochem. Bull. 19: 11–15.
- Flannery, M. L., F. J. G. Mitchell, S. Coyne, T. A. Kavanagh, J. I. Burke, N. Salamin, P. Dowdingand and T. R. Hodkinson. 2006. Plastid genome characterisation in Brassica and Brassicaceae using a new set of nine SSRs. Theor. Appl. Genet. 113: 1221–1231.
- Mizushima, U. 1980. Genome analysis in Brassica and allied genera. p. 89–105. In: S. Tsunoda, K. Hinata and C. Gomez-Campo (eds.). Brassica crops and wild allies: biology and breeding. Jpn. Sci. Soc. Press, Tokyo.
- Morinaga, T. 1934. Interspecific hybridization in Brassica VI. The cytology of F1 hybrids of B. juncea and B. nigra. Cytologia 6: 62–67.
- Nakamura, I. 2010. Methods of identifying eukaryotic species. JP2010088398.
- Nishi, S. 1980. Differentiation of Brassica crops in Asia and the breeding of ‘Hakuran’, a newly synthesized leafy vegetable. p. 133–150. In: S. Tsunoda, K. Hinata and C. Gomez-Campo (eds.). Brassica crops and wild allies: biology and breeding. Jpn. Sci. Soc. Press, Tokyo.
- Okamoto, S., M. Odashima, R. Fujimoto, Y. Sato, H. Kitashiba and T. Nishio. 2007. Self-compatibility in Brassica napus is caused by independent mutations in S-locus genes. Plant J. 50: 391–400.
- Organisation for Economic Cooperation and Development (OECD). 2012. Consensus document on the biology of the Brassica crops (Brassica spp.). OECD, Paris, France.
- Palmer, J. D., C. R. Shields, D. B. Cohen and T. J. Orton. 1983. Chloroplast DNA evolution and the origin of amphidiploid Brassica species. Theor. Appl. Genet. 65: 181–189.
- Prakash, S. and K. Hinata. 1980. Taxonomy, cytogenetics, and origin of crop Brassica, a review. Opera Bot. 55: 1–57.
- Rai, B., H. Takahashi, K. Kato, Y. I. Sato and I. Nakamura. 2012. Single-copy nuclear PolA1 gene sheds light on the origin of S genome with relationships to B and G genomes of polyploid wheat species. Genet. Resour. Crop Evol. 59: 1713–1726.
- Saitou, N. and M. Nei. 1987. The Neighbor-Joining method; a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4: 406–425.
- Song, K. M., T. C. Osborn and P. H. Williams. 1988. Brassica taxonomy based on nuclear restriction fragment length polyohisms (RFLPs) I. Genome evolution of diploid and amphidiploid species. Theor. Appl. Genet. 75: 784–794.
- Takahashi, H., B. Rai, K. Kato and I. Nakamura. 2010. Divergent evolution of wild and cultivated subspecies of Triticum timopheevii as revealed by the study of PolA1 gene. Genet. Resour. Crop Evol. 57: 101–109.
- Takahashi, H., T. Sato, Y-I. Sato and I. Nakamura. 2009. Genome-type-specific variation of the19th intron sequence within the RNA polymerase I largest subunit gene in the genus Oryza. Plant Syst. Evol. 282: 21–29.
- Takahata, Y. and K. Hinata. 1986. Application of a pattern analysis (qualification method III) for the isozyme relations in crop Brassicas. Cruciferae Newslett. 11: 12–13.
- Tamura, K., D. Peterson, N. Peterson, G. Stecher, M. Neiand and S. Kumar. 2011. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28: 2731–2739.
- Thompson, J. D., D. G. Higgins and T. J. Gibson. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl. Acids Res. 22: 4673–4680.
- Town, C. D., F. Cheung, R. Maiti, J. Crabtree, B. J. Haas, J. R. Wortman, E. E. Hine, R. Althoff, T. S. Arbogast, L. J. Tallon, M. Vigouroux, M. Trick and I. Bancroft. 2006. Comparative Genomics of Brassica oleracea and Arabidopsis thaliana reveal gene loss, fragmentation, and dispersal after polyploidy. Plant Cell 18: 1348–1359.
- U, N. 1935. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Japan. J. Bot. 7: 389–452.
- Vaughan, J. G. 1977. A multidisciplinary study of the taxonomy and origin of Brassica crops. BioScience 27: 35–40.
- Wang, X., H. Wang, J. Wang, R. Sun, J. Wu, S. Liu, Y. Bai, J. H. Mun, I. Bancroft, F. Cheng, S. Huang, X. Li, W. Hua, J. Wang, X. Wang, M. Freeling, J. C. Pires, A. H. Paterson. B. Chalhoub, B. Wang, A. Hayward, A. G. Sharpe, B. S. Park, B. Weisshaar, B. Liu, B. Li, B. Liu, C. Tong, C. Song, C. Duran, C. Peng, C. Geng, C. Koh, C. Lin, D. Edwards, D. Mu, D. Shen, E. Soumpourou, F. Li, F. Fraser, G. Conant, G. Lassalle, G. J. King, G. Bonnema, H. Tang, H. Wang, H. Belcram, H. Zhou, H. Hirakawa, H. Abe, H. Guo, H. Wang, H. Jin, I. A. Parkin, J. Batley, J. S. Kim, J. Just, J. Li, J. Xu, J. Deng, J. A. Kim, J. Li, J. Yu, J. Meng, J. Wang, J. Min, J. Poulain, J. Wang, K. Hatakeyama, K. Wu, L. Wang, L. Fang, M. Trick, M. G. Links, M. Zhao, M. Jin, N. Ramchiary, N. Drou, P. J. Berkman, Q. Cai, Q. Huang, R. Li, S. Tabata, S. Cheng, S. Zhang, S. Zhang, S. Huang, S. Sato, S. Sun, S. J. Kwon, S. R. Choi, T. H. Lee, W. Fan, X. Zhao, X. Tan, X. Xu, Y. Wang, Y. Qiu, Y. Yin, Y. Li, Y. Du, Y. Liao, Y. Lim, Y. Narusaka, Y. Wang, Z. Wang, Z. Li, Z. Wang, Z. Xiong and Z. Zhang. 2011. The genome of the mesopolyploid crop species Brassica rapa. Nature Genet. 43: 1035–1039.
- Zhang, X., H. Takahashi, I. Nakamura and M. Mii. 2008. Molecular discrimination among taxa of Petunia axillaris complex and P. integrifolia complex based on PolA1 sequence analysis. Breed. Sci. 58: 71–75.