ORIGINAL ARTICLES
Isolation and Characterization of S-RNase-homologous Genes Expressed in Styles in ‘Hyuganatsu’ (Citrus tamurana hort. ex Tanaka)
2019 Volume 88 Issue 3 Pages 338-346

Details
2019 Volume 88 Issue 3 Pages 338-346
In this study, S-RNase-homologous genes expressed in styles were isolated from a gametophytic self-incompatible citrus cultivar, ‘Hyuganatsu’. Sweet orange and clementine genome databases were searched to identify 13 ribonuclease T2 (T2 RNase) genes. Further blast searches using citrus EST databases were conducted with these 13 sequences as queries to obtain five additional EST sequences. Known T2 RNase genes, including the S-RNases of Rosaceae, Solanaceae, and Plantaginaceae were retrieved from the public database. All data collected from the databases were combined to make a dataset for phylogenetic analysis. From the phylogenetic tree, 10 citrus sequences were found to be monophyletic to the clade of S-RNases. Degenerate primers were designed from these genes to identify similar sequences in cDNA derived from ‘Hyuganatsu’ styles. RT-PCR and 3' and 5' RACE resulted in the isolation of three S-RNase-like sequences; these sequences were further characterized. Pistil-specific gene expression was confirmed for all sequences by RT-PCR; Citrus tamurana RNase1 (CtRNS1) and RNase3 (CtRNS3) had basic pI values (> 8) and molecular weights of approximately 25 kDa, consistent with S-RNase features of other families. The distribution of CtRNS1, CtRNS2, and CtRNS3 was investigated using 10 citrus cultivars. CtRNS1 and CtRNS2 did not correspond to any S-haplotypes in the 10 cultivars, while CtRNS3 matched the citrus’ S1-haplotype.
Citrus is the leading fruit crop worldwide. Some Citrus species display self-incompatibility (SI, to which the adjective self-incompatible is also abbreviated) (Yamamoto et al., 2006, 2012). In terms of fruit production, SI combined with parthenocarpy enables the production of the seedless fruit demanded by most consumers. However, SI in the absence of parthenocarpy results in the production of seedy fruit. Furthermore, production costs and labor inputs are higher because of the need for mixed planting with a compatible cultivar as a pollinizer and/or artificial pollination for stable fruit set. Therefore, understanding the mechanism regulating citrus SI has long been of interest with the objective of breeding seedless cultivars with improved fruit setting.
Citrus SI is of the gametophytic self-incompatibility (GSI) type. However, the key genes that determine S haplotypes and function in self/non-self recognition have not yet been identified. Among GSI plant species, the Rosaceae, Solanaceae, and Plantaginaceae families share the same SI system and the molecular mechanisms regulating this process have been better characterized (Matsumoto and Tao, 2016; Tao and Iezzoni, 2010). The SI system in these families is ruled by two different closely linked genes that define the pistil and pollen S specificities. The female S determinant is S-RNase which is homologous to Aspergillus RNase-T2 (T2 type RNase) according to homology-based sequence analysis (McClure et al., 1989). Thus, this system is commonly known as S-RNase-based GSI, and major fruit crops in the genera Malus, Pyrus, and Prunus exhibit S-RNase-based GSI, leading us to speculate that S-RNase-based SI may also occur in Citrus species.
Therefore, in this study we attempted to isolate style-expressed S-RNase-like genes in ‘Hyuganatsu’ (Citrus tamurana hort. ex Tanaka). ‘Hyuganatsu’ is a late cultivar that was identified in Miyazaki Prefecture, Japan, approximately 150 years ago and is now grown commercially in several prefectures, including Miyazaki, Kochi, and Shizuoka. ‘Hyuganatsu’ is an SI cultivar with a very low level of parthenocarpy (Miwa, 1951; Yamashita, 1978), resulting in seedy fruit that are contrary to consumer preference. Although similar attempts have previously been made to isolate genes homologous to Rosaceae, Solanaceae, and Plantaginaceae S-RNases from some Citrus species (Chai et al., 2011; Liang et al., 2017; Miao et al., 2011), no confirmed S-RNases have been identified in citrus to date (Zhang et al., 2018). However, recent technological advances in DNA sequencing and successful completion of citrus genome sequencing projects have resulted in the public availability of annotated genome sequences of several citrus species (Shimizu et al., 2017; Wu et al., 2014; Xu et al., 2013) that will facilitate the identification of homologous genes. We therefore used sequence information for T2 RNase genes from the clementine (C. clementina hort. ex Tanaka) and sweet orange (C. sinensis (L.) Osb.) genome databases to identify genes phylogenetically close to S-RNase genes. Degenerate primers were then designed using these genes to isolate style-expressed T2 RNases in ‘Hyuganatsu’. Putative T2 RNase genes were further characterized by predicting the isoelectric point of the encoded enzyme, determining the intron position and number, and investigating tissue-specific gene expression patterns. Furthermore, we assessed whether these features are consistent with the common features of S-RNase genes.
Field-grown Citrus tamurana hort. ex Tanaka ‘Hyuganatsu’ trees of the Laboratory of Pomology, University of Miyazaki were used in this study. Flowers at the balloon stage were collected in the 2013 flowering season and floral organs (stigma, style, ovary, petal, sepal, anther, and filament) were dissected. Mature fruit were also harvested to obtain the albedo, flavedo, and juice sac. Additionally, young leaves of the 10 diploid cultivars listed in Table 1 were collected. Cultivars that are self-incompatible, have at least one determined S haplotype, or have an apparent parent-progeny relationship were selected for the study (Table 1). All samples were collected in the field belonging to the Laboratory of Pomology, with the exception of ‘Nomabeni Hassaku’, ‘Sweet Spring’, and ‘Okitsu Wase’, which were collected at the Field Science Center, Faculty of Agriculture, University of Miyazaki. After collection, all samples were immediately frozen with liquid nitrogen and stored in a −80°C freezer for later use.

Details of the citrus cultivars examined in this study.
Genes annotated as ribonuclease T2 family members were identified from sweet orange (v1.1) and clementine (v0.9) genome databases in Phytozome (http://www.phytozome.net/) by using “ribonuclease T2” or “GO:0033897”, which is a term accession number assigned to the GO term “ribonuclease T2 activity”, as the search keywords. After T2 RNase sequences were obtained, similar sequences were also identified in the citrus EST database using tblastx in NCBI. Sequence data of T2 RNase genes in other angiosperms, which included S-RNases in Rosaceae, Solanaceae, and Plantaginaceae, were retrieved from DDBJ/EMBL/GenBank databases (Suppl. Table 1). An additional T2 RNase gene from Physcomitrella patens was obtained through Phytozome to act as an outgroup sequence. All amino acid sequences were aligned with ClustalW and a phylogenetic tree was constructed with the Neighbor-Joining method (Saitou and Nei, 1987) using a JTT substitution model in MEGA7 (Kumar et al., 2016).
Cloning of full-length sequences of T2/S-RNase-like genesIn the constructed phylogenetic tree, sequences from Citrus species that were monophyletic to the clade consisting of Rosaceae, Solanaceae, and Plantaginaceae S-RNases were selected. Their coding sequences (CDS) were aligned using ClustalW and degenerate primers were designed in the regions where nucleotide sequences were relatively conserved (Table 2; Suppl. Fig. 1).
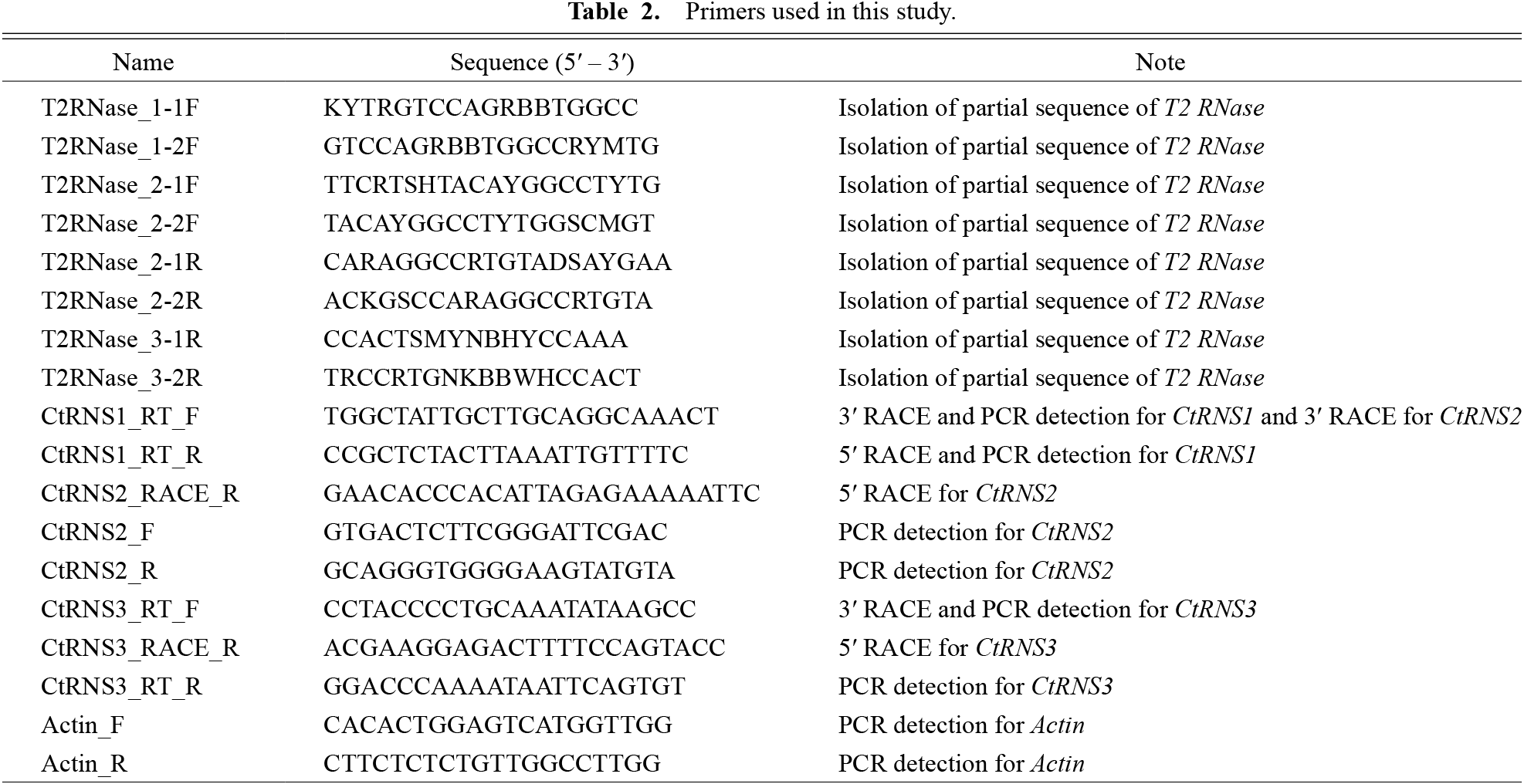
Primers used in this study.
Total RNA was extracted from about 300 mg of style tissue using RNAiso Plus (TaKaRa Bio, Shiga, Japan) followed by cDNA synthesis using 1 μg of total RNA with a SMARTerTM PCR cDNA Synthesis Kit (Clontech, Mountain View, CA, USA). Double-stranded cDNA was generated using PCR amplification according to the manufacturer’s instructions. PCR was then conducted using double-stranded cDNA as a template and degenerate primers, followed by nested PCR as required. Amplified fragments were cloned with the pGEM T-Easy vector (Promega, Madison, WI, USA) and Escherichia coli DH5α cells (Nippon Gene, Tokyo, Japan). Positive cells were selected, plasmids were extracted, and inserted fragments were sequenced.
Three different sequences were obtained from the sequencing results. Gene-specific primers were designed for these sequences and 5' and 3' RACE was carried out using a SMART RACE kit (Clontech) with 1 μg of total RNA extracted from the styles. Amplified fragments were cloned and sequenced as described above to obtain the full-length cDNA sequence. Deduced amino acid sequences, which were inferred from the longest ORF, combined with the dataset were used to construct the phylogenetic tree. Using the new dataset, the phylogenetic tree was reconstructed with the same method and settings.
Sequencing the genomic DNA to identify the intron number and locationGenomic DNA was extracted from the ‘Hyuganatsu’ leaves using the CTAB method (Doyle and Doyle, 1987). Extracted DNA concentrations were measured with a NanoDrop 2000 (ThermoFisher Scientific, Waltham, MA, USA) and adjusted to 10 ng·μL−1. Four primer sets were designed according to the full-length cDNA sequences for each T2 RNase; however, the forward primers for CtRNS1 and CtRNS2 were common because of the high similarity of these sequences (Table 2). PCR reactions were conducted using Ex Taq DNA polymerase (TaKaRa Bio). Amplified fragments were treated with ExoSAP-IT (ThermoFisher Scientific) and then direct-sequenced. Intronic regions were identified by comparing two sequences of gDNA and cDNA by ClustalW with the default parameter set in GenomeNet (https://www.genome.jp/tools-bin/clustalw).
Estimation of molecular weight, isoelectric point, motifs, and signal peptidesThe molecular weight and isoelectric point (pI) of the proteins identified from the sweet orange and clementine databases and those obtained from ‘Hyuganatsu’ style RNA were calculated using the ProtParam tool at ExPASy (Gasteiger et al., 2005). Motif searching was conducted using InterProScan (https://www.ebi.ac.uk/interpro/search/sequence-search). Signal peptide regions and subcellular localization were predicted using the SignalP 4.1 server (http://www.cbs.dtu.dk/services/SignalP/) and DeepLoc-1.0 (http://www.cbs.dtu.dk/services/DeepLoc/), respectively (Armenteros et al., 2017; Petersen et al., 2011).
Expression analysis using RT-PCRTotal RNAs were extracted from floral organ and fruit tissue with RNAiso Plus (TaKaRa Bio). First strand cDNA was synthesized using ReverTra Ace® qPCR RT Master Mix with gDNA Remover (Toyobo, Tokyo, Japan) in 10 μL reaction volumes containing 200 ng total RNA according to the manufacturer’s instructions. After synthesis, reaction solutions were diluted with the addition of 90 μL of TE buffer. PCR was performed using these cDNAs with EmeraldAmp® MAX PCR Master Mix (TaKaRa Bio) and 30 cycles of 95°C for 30 s, 57°C for 30 s, and 72°C for 30 s. The gene-specific primers used for reactions are shown in Table 2. Amplicons were stained with EtBr and visualized under UV after separation with 1% agarose gel electrophoresis.
Distribution of CtRNS1, CtRNS2, and CtRNS3 in other citrus cultivarsGenomic DNA of the citrus cultivars listed in Table 1 was extracted using the CTAB method (Doyle and Doyle, 1987) followed by purification via phenol/chloroform extraction. DNA solutions were quantified by NanoDrop 2000 and their concentrations were adjusted to 10 ng·μL−1. For the detection of CtRNS1 to CtRNS3 in the different cultivars, PCR amplification was carried out with EmeraldAmp® MAX PCR Master Mix (TaKaRa Bio) and primer sets for CtRNS1 to CtRNS3 (Table 2). Thermal cycling was as follows: 94°C for 1 min for initial denaturation and then 30 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s followed by a final extension at 72°C for 2 min. Amplicons were electrophoresed on a 1.5% agarose gel, stained with EtBr and visualized under UV light.
Six and seven genes annotated as ribonuclease T2 genes were identified from the sweet orange and clementine genome databases, respectively, using database searches (Table 3). Liang et al. (2017) used a similar approach to obtain T2 RNase genes by surveying different databases from those used in this study; they obtained seven and nine T2 RNase genes from the clementine v.1.0 and sweet orange v.2.0 databases, respectively. In this study, obtained amino acid sequences were queried using a tblastx search of the EST database that resulted in five hits. The corresponding sequence data were combined with the other T2 RNase dataset for the phylogenetic analysis. The phylogenetic tree indicated that 10 sequences originating from Citrus species made a cluster and were grouped in the same clusters as Rosaceae, Solanaceae, and Plantaginaceae S-RNase genes (Fig. 1). The nucleotides of seven of these sequences were re-aligned and six degenerate primers were designed (Table 2; Suppl. Fig. 1). After RT-PCR trials using a number of forward and reverse degenerate primer combinations, RT-PCR with the combination of T2RNase_1-1F and T2RNase_3-2R, followed by nested PCR using the primers T2RNase_1-2F and T2RNase_3-2R, finally resulted in the isolation of three different sequences; their full-length cDNA sequences were obtained by 3' and 5' RACE. These sequences were named CtRNS1 to CtRNS3 and were deposited in DDBJ/ENA/GenBank with accession numbers (LC408957, LC408958, and LC408959 for CtRNS1, CtRNS2, and CtRNS3, respectively).

Characteristics of T2 RNase genes found in clementine and sweet orange genome databases and style-expressed T2 RNase genes isolated from ‘Hyuganatsu’ in this study.

Phylogenetic tree of T2 RNase genes constructed by the NJ method. The newly isolated genes, CtRNS1 to CtRNS3, are indicated in bold. Class I to III beside the bars at the right of the tree indicates the T2 RNases classification based on Igic and Kohn (2001). Bracketed sequences in the citrus cluster except for CtRNS1 to CtRNS3 were realigned for the design of degenerate primers.
Their longest ORFs were considered to represent their deduced amino acid sequences. The lengths of the deduced amino acid sequences were 225, 113, and 219 for CtRNS1, CtRNS2, and CtRNS3, respectively (Table 3). The phylogenetic tree including CtRNS1 to CtRNS3 demonstrated that all CtRNS genes clustered with other citrus S-RNase-like sequences and were positioned within the S-RNase clades of Rosaceae, Solanaceae, and Plantaginaceae (Fig. 1). According to the previous phylogenetic analysis of T2 RNase, the S-RNase-based SI system evolved only once 120 million years ago before the split of the Asteridae and Rosidae (Igic and Kohn, 2001; Steinbachs and Holsinger, 2002; Vieira et al., 2008; Wikstrom et al., 2001). The order Sapindales, which includes the genus Citrus of the family Rutaceae, diversified 80–84 million years ago (Wikstrom et al., 2001). The appearance of Citrus occurred after the evolution of S-RNases; this may support our hypothesis that SI in Citrus is potentially regulated by S-RNase.
Characterization of CtRNS1 to CtRNS3 sequencesThe InterProScan search results verified that all the amino acid sequences from CtRNS1 to CtRNS3 contained the T2 RNase motif. T2 RNases, including S-RNases, have two sequences forming the active site that each contain histidine residues essential for catalytic activity (Kawata et al., 1990; Parry et al., 1997; Royo et al., 1994). CtRNS2, however, lacks one of the two active sites with the histidine residue, suggesting that CtRNS2 may not be a functional enzyme. A tblastn search of CtRNS1 to CtRNS3 in the clementine and sweet orange genome databases indicated that CtRNS1 showed more than 90% similarity to clementine0.9_032811m (98.7% identical and positive) and orange1.1g042583m (90.22% identical and positive), while CtRNS3 was similar to orange1.1g044555m with lower similarity (41.55% identical and 48.31% positive). These results are reflected in the phylogenetic relationship shown in Figure 1. SignalP searches indicated that the signal peptide sequence for extracellular localization located at the N-terminal was predicted in all three sequences. Additionally, the DeepLoc program predicted that all the proteins are likely to be soluble and localized in the extracellular space (Table 4). This is also a common feature of S-RNases (Anderson et al., 1989; Certal et al., 1999) and is thought to be associated with the nature of the interaction between S-RNase and pollen (Matsumoto and Tao, 2016), suggesting that CtRNS genes are correlated with SI in Citrus.

Likelihood of protein subcellular localization and type.
The predicted pI values and molecular weights of CtRNS1, CtRNS2, and CtRNS3 were 8.67 and 25.4 kDa, 6.98 and 12.5 kDa, and 8.01 and 25.4 kDa, respectively, determined using ProtParam (Table 3). S-RNase varied in terms of molecular weight (~22–35 kDa) and isoelectric point (8–10) (Roalson and McCubbin, 2003). According to the classification of the T2 RNase gene family by Igic and Kohn (2001), S-RNases are included in the Class III cluster. The proteins coded by Class III T2 RNases have basic isoelectric points (pI), while most T2 RNases are acidic (Irie, 1999; Ramanauskas and Igic, 2017). Ramanauskas and Igic (2017) did an analysis using a large dataset of T2 RNase genes including S-RNases and revealed that the median predicted pI value in Class I proteins was 5.04, 5.90 in Class II, and was more basic in Class III; the median pI of non-S-RNases was 8.56 and that of S-RNase was 9.18. The pI appears to be significantly correlated with S-RNase function, although the functional causes of the association of protein pI values are unclear (Ramanauskas and Igic, 2017). In the T2 RNases isolated in this study, CtRNS1 and CtRNS3 satisfied the pI and molecular weight ranges observed for S-RNase; the CtRNS2 protein molecule was too small and its pI was more acidic than the others.
Intron number and positionThe genome sequencing revealed that single introns of 320 bp and 375 bp were inserted at seemingly the same position in CtRNS1 and CtRNS3, respectively. While CtRNS2 had no introns, a comparison with the CtRNS1 genomic sequence indicated that CtRNS2 was identical to the sequence from the first exon to the middle of the CtRNS1 intron, suggesting that CtRNS1 and CtRNS2 are the same gene and that CtRNS2 may be an isoform or splicing variant of CtRNS1. The number of introns is one common feature of S-RNase lineage genes, with the exception of S-RNases in Prunus species. S-RNase genes other than those from Prunus species have only one intron in the HV region (Igic and Kohn, 2001; Tao and Iezzoni, 2010; Tao et al., 1999). Prunus S-RNases have an additional intron between the signal peptide and the start of the mature protein that was likely added after the separation of Prunus (Morimoto et al., 2015). Thus, the results here implied that CtRNS1, CtRNS2, and CtRNS3 have the same origin as the S-RNase-lineage genes in Rosaceae, Solanaceae, and Plantaginaceae species.
Tissue-specific expressionAll reported S-RNases are specifically expressed in the pistil, and especially in the style (Anderson et al., 1986; Tao et al., 1999; Xue et al., 1996; Yamane et al., 1999). RT-PCR results demonstrated that expression of CtRNS1 and CtRNS3 was restricted to the stigma, style, and ovary, and CtRNS2 showed strong expression in the pistil (Fig. 2). Although a non-S-RNase was also specifically expressed in the style in Prunus (Morimoto et al., 2015; Yamane et al., 2003), pistil-specific expression is a common feature of S-RNases and all three genes isolated in this study satisfied this feature.

RT-PCR analysis using various tissues of ‘Hyuganatsu’. RT-PCR was performed on stigma (Stg), style (Sty), ovary (Ov), anther (An), filament (Fil), petal (Pet), sepal (Sep), albedo (Al), flavedo (Fl), and juice sac (Js). M denotes a DNA marker (Gene Ladder 100; Nippon Gene, Tokyo, Japan).
Some T2 RNase genes in citrus are reportedly involved in fruit development (Liang et al., 2017). Liang et al. (2017) found that one gene (CsRNS1) out of nine T2 RNase genes was continuously up-regulated in sweet orange fruit from flowering to 240 d after flowering, suggesting that the S-like RNase genes obtained here may be involved in fruit development. However, none of the T2 RNase genes isolated in this study were expressed in fruit tissues, indicating that their function is not associated with fruit development. Their non-involvement in fruit development is also supported by the distant phylogenetic position of CtRNS1–CtRNS3 relative to the Class II clade. Most Class II T2 RNase genes are constitutively expressed, suggesting that they may have housekeeping roles (MacIntosh, 2011); the citrus T2 RNase genes related to fruit development and ovary senescence also belong to this clade (Chai et al., 2011; Liang et al., 2017).
Distribution of CtRNS1 and CtRNS3 in other citrus cultivarsAlthough it is possible that citrus SI is not governed by S-RNases, CtRNS1 and CtRNS3 are equipped with features that are similar to those of S-RNases from other families. Therefore, we investigated correlations between CtRNS1–3 and previously identified S haplotypes. In a survey of CtRNS1, CtRNS2, and CtRNS3 distributions in 10 citrus cultivars, CtRNS1 and CtRNS2 were detected in all cultivars tested, which supports the idea that CtRNS1 and CtRNS2 are the same gene and may not be involved in SI mechanisms, while CtRNS3 was found only in S1 haplotype-possessing cultivars, such as ‘Hyuganatsu’, ‘Tosabuntan’, ‘Banpeiyu’, and ‘Haruka’ (Fig. 3). Kim et al. (2011) defined the SI genotype (S genotype) of ‘Banpeiyu’ as S1S2 and tried to determine the S genotypes of some citrus cultivars using homozygous (S1S1 or S2S2) seedlings derived from a bud pollination of selfed ‘Banpeiyu’. ‘Hyuganatsu’ and ‘Tosabuntan’ reportedly contain the S1 allele because they were incompatible with the pollen grains of S1 homozygous progeny (Table 1; Kim et al., 2011). ‘Haruka’ is a chance seedling of ‘Hyuganatsu’ and its pollen parent was recently identified as ‘Natsudaidai’ (Shimizu et al., 2016). Therefore, the S1-haplotype of ‘Haruka’ could be inherited from ‘Hyuganatsu’. Among the cultivars that do not contain CtRNS3, clementine, ‘Nomabeni Hassaku’, and ‘Okitsu Wase’ did not have an S1 allele (Kim et al., 2011). Because the CtRNS3 distribution matched its presence/absence on the S1 allele, it is plausible that CtRNS3 is the S1 allele in citrus.

Distribution of CtRNS1, CtRNS2, and CtRNS3 in 10 citrus cultivars (1: ‘Hyuganatsu’, 2: ‘Ariake’, 3: clementine, 4: ‘Sweet Spring’, 5: ‘Tosabuntan’, 6: ‘Seike’ Navel, 7: ‘Banpeiyu’, 8: ‘Haruka’, 9: ‘Nomabeni Hassaku’, 10: ‘Okitsu Wase’). M denotes a DNA marker (Gene Ladder 100).
While we cannot conclude at present that CtRNS3 is an S gene product, it displays features similar to those of S-RNase. If CtRNS3 is confirmed to be the S1-gene, the present data will make a big contribution to the breeding of seedless cultivars in citrus.
The authors would like to thank Mr. Yasuhiro Udatsu at the Field Science Center, Faculty of Agriculture, University of Miyazaki for providing us with ‘Nomabeni Hassaku’, ‘Sweet Spring’, and ‘Okitsu Wase’ leaf samples.