ORIGINAL ARTICLES
Cloning and Molecular Characterization of CfMYBs Associated with the Regulation of Methyl Jasmonate Biosynthesis in Cymbidium faberi
2020 Volume 89 Issue 5 Pages 593-601

Details
2020 Volume 89 Issue 5 Pages 593-601
Cymbidium is one of the largest orchid genera and it is famous for its ornamental, cultural, and economic value. Many Cymbidium species have an elegant flower fragrance, including C. faberi, C. goeringii, C. ensifolium, C. kanran, C. sinense, and so on. Although the components of the flower fragrance have been identified in many orchids, the molecular mechanism of their formation and regulation has not been explored. As one of the main components of flower fragrance, methyl jasmonate (MeJA) has been selected and the biosynthesis pathway was elucidated in some Cymbidium orchids. In order to modify the traits of flower fragrance in Cymbidium orchids, the underlying regulatory mechanisms need to be identified. In this study, four MYB transcription factors screened from RNA-seq results of C. faberi were successfully cloned and molecularly characterized. The activation of these CfMYBs with CfAOC and CfJMT promoters suggested that they may participate in the regulation of MeJA formation in C. faberi. This result could provide molecular support for the genetic modification and breeding of new C. faberi cultivars.
Cymbidium is the most important genus of so-called “oriental” orchids, which have an elegant flower fragrance, but lack the showy colors of most cultured tropical orchids. The flower fragrance of the main species of Cymbidium, including C. faberi, C. goeringii, C. ensifolium, C. kanran, and C. sinense, has been analyzed and its components were identified (Omata et al., 1990; Feng et al., 2009; Yang et al., 2011). Since a flower fragrance is generally made up of a blend of compounds and is quite volatile, it is difficult to demonstrate the biosynthetic pathway of flower fragrance at the molecular level. The genome sequence of Phalaenopsis equestris was released in 2014, but research on Cymbidium species has lagged behind (Cai et al., 2015). Furthermore, due to limited propagation and cultivation, most oriental orchids are directly transplanted from the wild, which severely damages the wild resources of Cymbidium. Therefore, it is necessary to clarify the biosynthetic pathways and regulatory mechanisms of the flower fragrance in Cymbidium to provide guidance for new approaches to genetic breeding.
Cymbidium faberi is a species with a very long cultivation history, and it has been extensively studied in terms of its population dynamics, development of molecular markers, identification of its flower fragrance components, and propagation optimization (Yang et al., 1994; Chen et al., 2005; Vendrame et al., 2007; Hossain et al., 2010, 2013; Tao et al., 2011). However, C. faberi was rarely the subject of research at the molecular level. The main components of the flower fragrance of C. faberi are methyl jasmonate (MeJA) and methyl epijasmonate, respectively, accounting for about 9.62% and 8.25% of the total volatile components, as verified by different teams (Omata et al., 1990; Feng et al., 2009; Zhou et al., 2018). The biosynthetic pathway of MeJA has been revealed in model plants, and the crucial enzymes have been cloned and functionally characterized. These include lipoxygenase (LOX), allene oxide synthase (AOS), allene oxide cyclase (AOC), 12-oxy-phytodienoic acid reductase (OPR) and jasmonic acid carboxyl methyltransferase (JMT) (Sasaki-Sekimoto et al., 2013).
Methyl jasmonate is formed from jasmonic acid (JA) in a reaction catalyzed by JMT, which is widely present in plants and participates in many plant developmental processes. In Arabidopsis, the three MYC transcription factors, MYC2, MYC3, and MYC4, are well known to be involved in the regulation of JA biosynthesis (Lorenzo et al., 2004; Cheng et al., 2011; Song et al., 2014). Another subgroup of MYC-like transcription factors, the JAMs, negatively regulate the JA pathway by competitively binding to the MYC target sequences (Fernández-Calvo et al., 2011; Nakata et al., 2013; Sasaki-Sekimoto et al., 2013, 2014). In general, these MYC transcription factors interact with MYB transcription factors to form a complex that is active in the fine regulation of downstream genes by binding to a G-box motif (Schweizer et al., 2013).
MYB transcription factors were first discovered in avian myeloblastosis virus, the causative agent of acute myeloblastic leukemia (Lüscher and Eisenman, 1990). In recent years, they were found to be one of the largest transcription factor families in plants, playing crucial roles in the regulation of many processes related to growth, development, physiological metabolism, cell morphogenesis and so on (Abe et al., 2013). Most MYBs comprise an N-terminal of the Myb domain that can be categorized into four classes: 1R-MYB/MYB-related, R2R3-MYB, 3R-MYB, and 4R-MYB. The R2R3 MYB transcription factors are arguably the most widely investigated, and were found to be involved in plant responses to biotic and abotic stresses, as well as the regulation of secondary metabolism. (Kranz et al., 2000). The N-terminus of MYBs is crucial for their binding to the specific target sequence, TAACTG, while the C-terminus often contains an activation domain to improve the expression of downstream genes (Biedenkapp et al., 1988; Kranz et al., 1998).
Recently, many MYBs have been applied to improve the traits of ornamental plants, such as flower color and fragrance (Colquhoun and Clark, 2011). Two R2R3-MYB genes, EOBII and ODO1, were cloned from Petunia hybrida and were found to participate in the regulation of the benzenoid pathway, which determines the floral aroma of petunias (Verdonk et al., 2005; Spitzer-Rimon et al., 2012). Similarly, the MYB transcription factor Pap1 (Production of Anthocyanin Pigment 1) was found to increase the production of volatile phenylpropanoid/benzenoid compounds 10-fold, as well as increasing the pigmentation of flowers (Ben Zvi et al., 2008). Overall, similar findings were reported in many other plants, including kiwifruit (Ampomah-Dwamena et al., 2019), purple tumorous stem mustard (Xie et al., 2019), strawberry (Delgado et al., 2018), Ginkgo biloba (Zhang et al., 2018), Populus tomentosa (Wang et al., 2019), and tomato (Jian et al., 2019), and it was also reported that some MYB transcription factors are involved in the formation of flower pigmentation and volatile organic compounds.
In order to investigate the biosynthesis and regulation of flower fragrance in C. faberi, we compared the gene expression in scented and non-scented flowers by RNA-seq, including the blooming stage and withered stage. Among 379 transcription factors screened among the differentially expressed genes, 55 were found to belong to the MYB family, which is the most abundant of all transcription factor families (Xu et al., 2019). In this study, we cloned four CfMYB genes from C. faberi that could activate the expression of CfAOC and CfJMT promoters in the MeJA biosynthetic pathway, suggesting that these CfMYBs may participate in the regulation of flower fragrance in C. faberi. This result provides theoretical support for the genetic breeding of new orchid cultivars.
The wild C. faberi was collected from the mountains of Dangyang, Hubei province, China (30°55'25"N, 111°51'24"E), and transplanted to the greenhouse of Wuhan University of Bioengineering in 2011. It often grows in the shade of trees at locations with a deep humus layer and avoids exposure to direct sunlight. C. faberi needs a moist growth environment, but generally needs less humidity than C. goeringii. In the greenhouse, the plantlets of C. faberi were cultivated in pots mainly containing clay particles, bark, vermiculite, perlite and plant ash. They were kept at a day/night temperature of 24°C under a 16 h light/8 h dark photoperiod. Sterilized plantlets of this material were harvested and stored in the climate chamber of the Center of Applied Biotechnology. The herbarium specimens were deposited at Jiangxi University of Traditional Chinese Medicine (Specimen ID: Y. Zhou & Y. Q Xu 20120301). The collection stages and tissues were reported previously (Xu et al., 2019). The total RNA extraction followed the protocol of the Trizol reagent kit (Takara Bio Inc., Japan). The first-strand cDNA was synthesized using a SMART II reverse-transcriptase kit (Takara Bio) and stored at −20°C for subsequent use.
Based on the results of RNA-seq, about 20 unigenes that were annotated to code for MYB transcription factors were screened out by bioinformatic analysis, including Unigene9303, Unigene80577, Unigene32267, Unigene46887, Unigene56756, Unigene39159-2, Unigene24370-1, Unigene11323, Unigene49145-2, Unigene20726-2, Unigene20569, Unigene40805-2, Unigene43551, Unigene61090, Unigene71641, Unigene49495-1, Unigene40504, Unigene36984, Unigene85481-1, and Unigene12759-1. Another 10 MYB proteins related to the MeJA biosynthesis pathway reported in other plants were downloaded from the National Center for Biotechnology Information (NCBI), including FaMYB1 (AF401220), FaMYB9 (JQ989281), FaMYB10 (MG456859), and FaMYB11 (JQ989282) from Fragaria × ananassa (Delgado et al., 2018), GsMYB15 (MH796674) from wild soybean (Shen et al., 2018), SlMYB14 (NM_001247333.1), and SlMYB75 (NM_001279063) from Solanum lycopersicum (Jian et al., 2019), as well as AtMYB14 (NM_128674), AtMYB102 (NM_118264.3), and AtMYB113 (NM_105308.2) from Arabidopsis thaliana (De Vos et al., 2006; Li et al., 2014). These MYBs were used to construct a phylogenetic tree using MEGA5.1 software.
Combined with the differentially expressed transcription factors between the blooming stage and the withered stage, four unigenes, Unigene20569, Unigene32267, Unigene9303, and Unigene24370, were selected for cloning of full-length coding sequences. The primers were designed at the initial codon position and the terminal codon position based on the prediction of the open reading frame, as listed in Table S1. The upregulated MYBs unigenes were amplified using the cDNA from the blooming stage as a template, and the down-regulated MYB unigenes were amplified with the cDNA from the withered stage as a template. The PCR reaction system comprised 1 μL cDNA templates, 2 μL dNTP mix (2.5 mM each), 2 μL 10 × Ex Taq buffer, 0.1 μL Ex Taq enzyme, 0.5 μL forward and reverse primers (10 mM), and ddH2O up to a volume of 20 μL. All of the reagents were purchased from Takara Bio. The PCR temperature program encompassed 35 cycles of 94°C for 30 s, 53°C for 30 s, and 72°C for 1 min each. The target PCR products were ligated into the pMD18-T vectors and sequenced at Sangon Biotech (China).
Bioinformatic analysis of CfMYBs and construction of a phylogenetic treeThe molecular mass and pI of the putative encoded CfMYB proteins were predicted using ProtParam (<http://web.expasy.org/protparam/>). The subcellular localization of CfMYBs was predicted using CELLO v.2.5 (<http://cello.life.nctu.edu.tw/>). The conserved domains of the CfMYBs were analyzed using the conserved domains database (CDD) from NCBI (<http://www.ncbi.nlm.nih.gov/cdd>). A phylogenetic tree of CfMYB and other MYB proteins related to the MeJA biosynthesis pathway derived from Fragaria × ananassa, wild soybean, Solanum lycopersicum and Arabidopsis thaliana was constructed using MEGA5.1 software in order to identify the homology relationships of the MYB proteins.
Analysis of the spatio-temporal expression patterns of the four CfMYB genes by qRT-PCRThe expression patterns of the CfMYB genes in the root, leaf, and flower tissues of C. faberi were determined by qRT-PCR. The flower samples were separated into early flowering stage, blooming stage and withered stage samples. The gene-specific primers for unigenes were named qUnigene20569-F1, qUnigene20569-R1, qUnigene32267-F1, qUnigene32267-R1, qUnigene9303-F1, qUnigene9303-R1, qUnigene24370-F1, and qUnigene24370-R1, as listed in Table S1. The β-actin gene was used as the internal control. SYBR green dye (Thermo Fisher Scientific, USA) was used for fluorescence signal determination and dissociation curves were applied to detect primer dimers and other non-specific by-products. The PCR procedure was performed at 95°C for 10 min, followed by two steps of 95°C for 15 s and 60°C for 1 min (ABI 7500; Applied Biosynthesis, USA). The relative expression value was calculated using the 2−ΔΔCT (Livak) method as follows: ΔΔCT = (CT, gene- CT, actin) samples- (CT, gene- CT, actin) control. The samples were analyzed in biological triplicates.
Construction of expression vectors for a dual-luciferase assayIn order to assess whether the CfMYB proteins are involved in the MeJA biosynthesis pathway, the identified CfMYB unigenes were cloned into the pGreen II 62-SK vector by restriction digestion. Unigene9303, Unigene20569, and Unigene32267 were amplified using primers containing XbaI and KpnI digestions sites, and cloned to produce pGreen II 62-SK-9303, pGreen II 62-SK-201569, and pGreen II 62-SK-32267, respectively. Similarly, Unigene24370 was cloned using XbaI and BamHI digestions sites, resulting in pGreen II 62-SK-24370. The effector vectors were constructed based on the pGreenII Luc-0800 with CfAOC and CfJMT promoters, derived from the MeJA biosynthesis pathway of C. faberi. The recombinant plasmids were all verified by digestion with corresponding restriction enzymes and sequenced at Sangon Biotech.
Dual-luciferase assay to assess the regulatory effects of CfMYBs in transiently transformed tobacco plantsThe recombinant plasmid pGreen II 62-SK-CfMYBs was respectively co-transformed into Agrobacterium tumefaciens GV3101 with the pSOUP helper plasmid. Then, the bacteria were harvested and mixed with the Agrobacterium culture transformed with a pGreenII Luc-0800-CfAOC/CfJMT reporter plasmid. The mixture was resuspended in an infiltration buffer containing 10 mM MgCl2 and 0.5 μM acetosyringone (Sangon Biotech), which was then infiltrated into the abaxial sides of N. benthamiana leaves. Leaf discs of approximately 0.5 cm2 were collected after two days of infiltration for analysis using the Dual-Luciferase Reporter Assay System (Promega, USA) based on the ratio of firefly luciferase (Luc) driven by the effector vector to the renilla luciferase (Ren), driven by the 35S promoter (Luc/Ren). The samples were analyzed in biological triplicates.
Statistical analysisData on the relative expression levels of CfMYB genes and the relative ratios of RLU1/RLU2 are presented as the means ± SE from n = XY parallel experiments, and were subjected to analysis of variance using SigmaPlot 12.5 software.
Based on the results of RNA-seq, 27 MYBs were up-regulated and 28 were down-regulated in the blooming stage compared to the withered stage. About 20 of 55 unigenes of CfMYB transcription factors were selected to construct a phylogenetic tree with 10 MYB transcription factors reported to be involved in the metabolic pathways of MeJA in other plants. With the exception of Unigene36984, Unigene85481, and Unigene12759, the other 17 unigenes were closely related to other MYB transcription factors, and clustered together. Among them, Unigene 9303 was closely related to FaMYB9 and FaMYB11, and Unigene 32267 clustered together with FaMYB1, which was previously reported to be associated with JA-mediated responses in accumulation of proanthocyanidins in strawberry (Delgado et al., 2018). Unigenes 24370 and 20569 were separated into another subgroup (Fig. 1). According to the RNA-seq results, Unigenes 32267 and 20569 were up-regulated in flowers at the blooming stage, while Unigenes 9303 and 24379 were down-regulated. Therefore, these four CfMYB unigenes were selected for cloning from C. faberi. Fragments with lengths of about 750 bp, 750 bp, 1 kb, and 1.5 kb were amplified using the cDNA templates corresponding to Unigene 32267, 9303, 24370, and 20569, respectively (Fig. 2). Then, these PCR products were ligated into the T-vector for sequencing.

Phylogenetic analysis of CfMYB transcription factors derived from RNA-seq results and MYBs from other plants related to the MeJA pathway, including FaMYB1 (AF401220), FaMYB9 (JQ989281), FaMYB10 (MG456859), and FaMYB11 (JQ989282) from Fragaria × ananassa, GsMYB15 (MH796674) from wild soybean, SlMYB14 (NM_001247333.1) and SlMYB75 (NM_001279063) from Solanum lycopersicum, as well as AtMYB14 (NM_128674), AtMYB102 (NM_118264.3), and AtMYB113 (NM_105308.2) from Arabidopsis thaliana. The phylogenetic tree was constructed using MEGA5.1 software with a bootstrap test of phylogeny based on the Neighbor-Joining method with default parameters.

Gel electrophoresis of the full-length fragments encoding the four CfMYB unigenes. a. Unigene 32267; b. Unigene9303; c. Unigene24370; d. Unigene20569. M1: DL5000 DNA Marker; M2: DL2000 DNA Marker.
The sequences of Unigenes 32267, 9303, 24370, and 20569 were 759 bp, 777 bp, 903 bp, and 1656 bp in length, respectively, encoding proteins of 252, 258, 300, and 551 amino acids, with expected molecular masses of 28.9, 29.6, 33.3, 61.0 kDa, and isoelectric points (pI) of 8.79, 8.78, 8.47, 8.63 based on ProtParam analysis. According to Target P1.1 and CELLO v.2.5 analysis, these CfMYBs transcription factors were probably located in the nucleus. The conserved domains of the CfMYBs were analyzed using the CDD tool on the NCBI website, and these proteins were all characterized as members of the MYB super family.
The protein sequences of these four CfMYB transcription factors combined with 10 MYBs from other plants were subjected to multiple alignment using BioEdit software. The sequence alignment revealed that these CfMYBs contained typical R2 and R3 DNA-binding domains, as shown in Figure 3. These MYB proteins were used to construct a phylogenetic tree using a bootstrap test of phylogeny based on the Neighbor-Joining method with default parameters. As shown in Figure 4, the CfMYBs showed similar clustering to that obtained in the unigene screening, again suggesting that these four unigenes belong to the MYB super family.

Sequence analysis of the identified CfMYBs. Multiple sequence alignment of the deduced CfMYB proteins with MYB proteins from other plants using BioEdit software. Identical and similar amino acids are represented in the same background color. The R2 and R3 domains are indicated by lines above.

Phylogenetic analysis of four CfMYB proteins and other MYB transcription factors from different species related to MeJA metabolism. The phylogenetic tree was constructed using MEGA5.1 with a bootstrap test of phylogeny based on the Neighbor-Joining method with default parameters.
In order to investigate the expression patterns of the four CfMYB transcription factors in different organs and at different flowering stages, the relative gene expression levels of the CfMYBs in roots, leaves and flowers were quantified by qRT-PCR. The results showed that Unigenes 32267 and 20569 were highly expressed in flowers at the blooming stage, while Unigene32267 showed the lowest expression in flowers at the withered stage and Unigene20569 showed the lowest expression in the roots. Unigenes 9303 and 24370 were highly expressed in flowers at the withered stage, and their expressions were lowest in the early flowering stage (Fig. 5). These results were consistent with the RNA-seq analysis, which indicated that Unigenes32267 and 20569 were up-regulated, while Unigenes 9303 and 24370 were down-regulated, in flowers at the blooming stage compared to the withered stage.

The expression pattern of four CfMYB unigenes in Cymbidium faberi. Flower 1: flowers at the early flowering stage; Flower 2: flowers at the blooming stage; Flower 3: flowers at the withered stage.
In order to explore whether the identified CfMYBs are associated with the MeJA biosynthesis pathway, the coding sequences of the four unigenes were cloned into dual-luciferase detection vectors, which were then verified by restriction enzyme digestion and sequencing. The transfected tobacco leaves were collected and the relative luciferase ratios were measured using a fluorescence detector. The results showed that all four CfMYBs could increase the activity of CfAOC and CfJMT promoters, irrespective whether they were up- or down-regulated in the flowers of C. faberi (Fig. 6). All of the CfMYBs significantly increased the transcription activity of the CfJMT promoter more than that of the CfAOC promoter, suggesting that they all participate in the regulation of the MeJA pathway to some extent. Unigene 9303 exhibited significantly weaker effects than Unigene 24370 (Fig. 6b). When the up- and down-regulated CfMYBs were mutually combined to test their influence on the activities of promoter sequences, all showed elevated promoter activity, and the combinations with Unigene 9303 showed the least improvement (Fig. 6c). This clearly resulted in activation of the CfJMT promoter and suggested that CfMYB transcription factors may mutually influence their activating effects in the regulation of downstream genes. Sometimes, they may form a complex to exert their function on the target genes together.
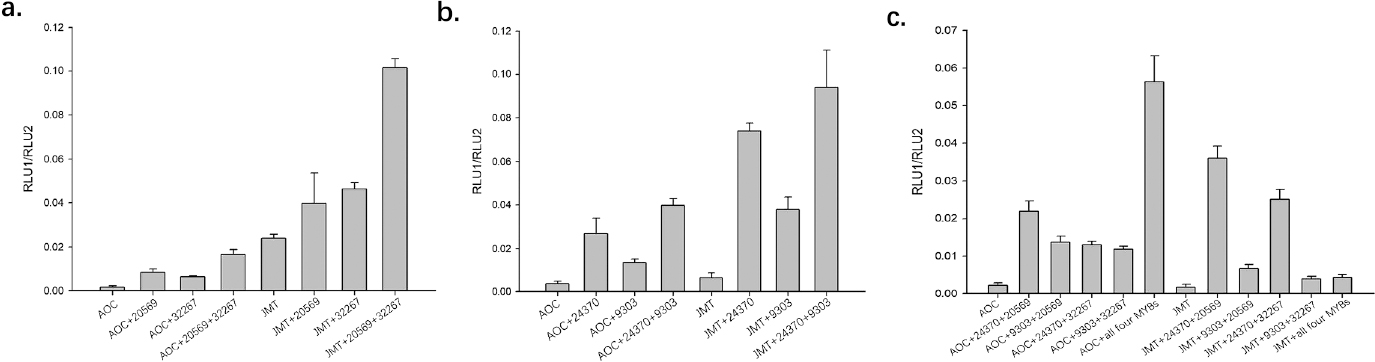
Dual-luciferase assay of the effect of the four CfMYB transcription factors on transcription from CfAOC and CfJMT promoter sequences. The LUC/REN ratio of the CfAOC and CfJMT promoter vectors plus pSOUP was used as a control. a. The LUC/REN ratio of the up-regulated CfMYB transcription factors in tobacco leaves co-transformed with CfAOC or CfJMT promoter vectors. b. The LUC/REN ratio of the down-regulated CfMYB transcription factors in tobacco leaves co-transformed with CfAOC or CfJMT promoter vectors. c. The LUC/REN ratio of combinations of the up- and down-regulated CfMYB transcription factors in tobacco leaves co-transformed with CfAOC or CfJMT promoter vectors.
C. faberi is an important horticultural plant, and its wild population has been listed as endangered for a long time. In order to preserve the wild resources of orchids and develop new directions for genetic breeding, it is necessary to first investigate the molecular mechanisms that control the economic traits, such as leaf shape, flower shape, flower color, and flower fragrance. We previously cloned many structural genes and transcription factors involved in the biosynthesis of flower fragrance in C. faberi. In this study, another four CfMYB transcription factor genes were successfully cloned and their relationship with MeJA biosynthesis was primarily explored. This suggested that these four CfMYB transcription factors may participate in the regulation of the MeJA pathway in C. faberi.
Based on the multiple alignment and phylogenetic tree analysis, these four CfMYB transcription factors belong to the R2R3-MYB subgroup, which is characterized by conserved R2 and R3 domains. They clustered together with other R2R3-type MYB subgroups from other plants, especially those involved in the metabolism of MeJA in other plants. The R2R3-MYB gene AtMYB102 was shown to respond to salt stress, ABA, JA, wounding and defense against insects or herbivores (De Vos et al., 2006). In the genome-wide screening, about 122 typical R2R3-MYB proteins were identified in tomato, of which 21 SlMYB genes were significantly affected by MeJA treatment (Li et al., 2016). It was reported that GhJAZ2’s interaction with the R2R3-MYB transcription factor GhMYB25-like could influence cotton fiber initiation and Arabidopsis trichome initiation through the regulation of JA homeostasis (Hu et al., 2016). Previous research also found that the overexpression of a peach R2R3-MYB transcription factor in tobacco could not only regulate flavonoid biosynthesis, but also up-regulate the JA biosynthesis and signaling pathways in flowers (Rahim et al., 2019). A JcMYB2 screened out by genome-wide identification from Jatropha curcas was significantly induced by cold, salt and MeJA, as well as slightly by ABA (Peng et al., 2016). The presence of two MeJA response elements in the promoter of GsMYB15 from wild soybean suggested that it may be involved in insect-plant interactions (Shen et al., 2018). Exogenous MeJA treatment or biotic and abiotic stresses generally give rise to variations in the endogenous JA pathway in plants, which are guided by an orchestra of transcription factors.
MYB transcription factors were also found to play a role in the formation of flower fragrance in plants. When the MYB transcription factor Production of Anthocyanin Pigment 1 (Pap1) was introduced into Petunia hybrida, it not only increased the pigmentation of flowers, but also dramatically increased the production of volatile compounds up to tenfold (Ben Zvi et al., 2008). In petunia, some other MYB transcription factors including ODORANT1 (ODO1), EMISSION OF BENZENOIDS I (EOBI), and EOB II, members of the R2R3-type MYB family, are crucial regulators of floral scent biosynthesis (Verdonk et al., 2005; Spitzer-Rimon et al., 2012). The overexpression of SlMYB75 in tomato led to the accumulation of anthocyanin and enhancement of volatile aroma production in the fruits (Jian et al., 2019). Methyl jasmonate is a major component of the C. faberi flower fragrance, and the CfMYB transcription factors identified in this study could regulate and activate the promoters of CfAOC and CfJMT, suggesting that they may also influence scent production in C. faberi.
It is well known that MYB transcription factors interact with bHLH transcription factors and a WD40 protein to form MBW complexes to exert their effect on downstream genes. It was shown that jasmonic acid promotes anthocyanin accumulation in Arabidopsis under light through the interaction of JAZ2 proteins and MBW complexes including MYB75 (Li et al., 2014). In strawberry, some specific MBW complex-related genes, such as FabHLH33 and FaMYB1/9/11, were up-regulated, while FabHLH3 and FaMYB10 were down-regulated, following the application of a JA-Ile biosynthesis inhibitor in MeJA treated fruits, which caused the accumulation of anthocyanin and proanthocyanidins in fruits (Delgado et al., 2018). In purple tumorous stem mustard, anthocyanins could be enhanced by sucrose and MeJA in sprouts under light conditions, and the expression of the MBW complex-related genes BjTT8, BjMYB1, BjMYB2, and BjMYB4 was correspondingly stimulated (Xie et al., 2019). In this study, two of the CfMYB transcription factors, Unigene20569 and Unigene32267, were up-regulated in flowers at the blooming stage, and the other two were down-regulated at this stage, as confirmed by both the RNA-seq and qRT-PCR results. However, these four transcription factors all had the same positive effects in the activation of the CfAOC and CfJMT promoters in the dual-luciferase assay. It was speculated that these four transcription factors work together with other transcription factors to exert their function on the target MeJA-related genes in C. faberi. However, a dual-luciferase detection experiment carried out in tobacco, in which only the MYB transcription factors were present, showed that their activation domains in the C-termini of MYB proteins could activate the expression of CfAOC and CfJMT promoters, but could not inhibit these promoters due to a lack of other transcription factor complex components.
In order to investigate the molecular mechanisms that control the biosynthesis of flower fragrance in C. faberi, many important genes were cloned to identify their functions in the MeJA metabolic pathway. In this study, another four MYB transcription factors involved in the MeJA pathway were cloned, and they were confirmed to enhance the activities of CfAOC and CfJMT promoters in transiently transfected tobacco plants. This result provides new information on the regulation of MeJA formation in C. faberi and supports the genetic modification of new cultivars, which can support the conservation of wild C. faberi resources. It also offers a reference for manipulating the flower fragrance traits in other non-scented orchids to increase their economic and ornamental value in the future.