Abstract
Portulaca umbraticola, an important ephemeral summer bedding plant, may be a model system for studies on flowering behavior under heat stress. Herein, we report the first comprehensive transcriptome analysis of P. umbraticola by RNA-sequencing. We generated 22.15 G bases which were assembled into 68,928 unigenes, and a total of 43,880 coding sequences (CDS) were detected. We also detected 13,603 simple sequence repeats on 10,784 unigenes, and predicted 1,444 transcription factor (TF) coding unigenes. Subsequently, we focused on gene discovery in the areas of flower senescence and opening rhythm. We identified the majority of transcripts involved in ethylene biosynthesis, perception, and signaling pathways, as well as transcripts corresponding to circadian clock components. Expression profiles of the ethylene biosynthesis and signaling pathway genes in two cultivars with different flower longevity showed a clear correlation with previously observed endogenous ethylene production. PuACS1 and PuACO2 transcripts showed higher expression, and peaked earlier in a short lived cultivar than in a long lived cultivar. Expression analysis of the core clock component genes showed PuCCA1/PuLHY and PuTOC1 under reciprocal circadian regulation similar to that found in Arabidopsis thaliana, and other plant species. This study provides the first steps in understanding the molecular mechanisms underlying flowering traits in P. umbraticola, and paves the way for future integrated insights into molecular genetics, biotechnology and physiological studies of P. umbraticola.
Introduction
The genus Portulaca has approximately 100 species distributed worldwide, mainly in the tropics and subtropics (Ocampo and Columbus, 2012). In the order Caryophyllales, Portulaca is the only known example of a dicot that has a C4 photosynthetic pathway (Voznesenskaya et al., 2010). The transition to a crassulacean acid metabolism (CAM) from C4 status was also reported in common purslane, Portulaca oleracea (Lara et al., 2004). Thus, plants in the genus Portulaca have a high tolerance to heat and drought stress.
Wingpod purslane, P. umbraticola, an annual summer bedding plant, has recently become an important part of the floriculture industry since the introduction of the ‘Wildfire mixed’ cultivar to the horticultural market (Matthews et al., 1992). P. umbraticola is an ephemeral flower which opens early in the morning and wilts in the afternoon; solar radiation and relatively high temperatures are essential for rapid and full opening (Ichimura and Suto, 1998). A lot of new cultivars of this species, with different flower shapes/colors, as well as flowering behavior, were recently bred (Fig. 1A). Recent studies reported cultivars that show differences in flower longevity produce different amounts of endogenous ethylene during petal senescence (Maguvu et al., 2016). Although the mechanism of ethylene response related to plant organ senescence, and the circadian clock system, which is vital for reproductive success, are well understood in the model plant Arabidopsis thaliana, little is known about the molecular mechanism underlying flower opening rhythm and senescence. Vase life was improved in plants that exhibit ethylene related senescence, such as carnations, Dianthus caryophyllus (Onozaki, 2018). The development of molecular genetic analysis tools such as whole genome sequences, and expressed sequence tags (ESTs), or higher density linkage maps have paved the way for molecular studies, as well as DNA marker assisted breeding (Kumari et al., 2013; Sreenvasulu et al., 2002). To the best of our knowledge, no such tools have been developed for P. umbraticola.

High-throughput DNA sequencing using next-generation sequencers (NGS), and particularly the development of ESTs, are cost effective and reliable methods for rapidly creating gene databases (Adams et al., 1993). These databases can be used in conjunction with bioinformatics tools to mine candidate genes that are involved in traits of interest. The large ESTs collections are also known to facilitate the design of microarrays, as well as the identification of single nucleotide polymorphisms (SNPs) and simple sequence repeats (SSRs). SNPs can be converted into genetic markers, and it is possible to use these markers to develop genetic maps (Gut, 2001; Rafalski, 2002). SSRs can also be used for genetic map construction (Morgante et al., 2002).
The ethylene biosynthesis and signaling pathways in plants are well characterized, and the genes encoding key enzymes were isolated (Kende, 1989). Ethylene is synthesized through the following pathway: methionine → S-Adenosyl methionine → 1-aminocyclopropane-1-carboxylic acid (ACC) → ethylene. The last two reactions are known to be the rate limiting steps, and are catalyzed by ACC synthase (ACS) and ACC oxidase (ACO), respectively. The ethylene is perceived by its receptors such as ETHYLENE RESPONSE SENSOR 1 (ERS1), ERS2, ETHYLENE RECEPTOR 1 (ETR1), ETR2, and ETHYLENE INSENSITIVE 4 (EIN4), which triggers a downstream signalling cascade involving CONSTITUTIVE TRIPLE RESPONSE 1 (CTR1), EIN2, EIN3, and ETHYLENE RESPONSE FACTORs (ERFs). In carnations, cultivars with a long vase life had lower ethylene production, which was also correlated with lower ACS and ACO expression than flowers with a short vase life (Tanase et al., 2008). An increase in ethylene production was shown to correlate with the expression levels of ACS and ACO genes (Park et al., 1992; Wang and Woodson, 1991). In this report, the expression patterns of ethylene biosynthesis, signaling, and perception genes were characterized in two P. umbraticola cultivars that vary in flower longevity and endogenous ethylene production.
The plant circadian clock regulates almost every aspect of plant growth, development, and reproduction. The mechanisms of the circadian clock are well characterized in the model plant A. thaliana (Sanchez and Kay, 2016). Tampering with clock-associated genes was shown to affect the rhythm of flower opening; silencing coyote tobacco, Nicotiana attenuata, LATE ELONGATED HYPOCOTYL (LHY), and ZEITLUPE (ZTL) alter circadian rhythms in flowers (Yon et al., 2016). A correlation between flower opening and the expression of circadian-clock associated genes in relation to the length of the dark period were reported in Ipomoea nil (Shinozaki et al., 2014).
In our previous study (Maguvu et al., 2018; Fig. 1B), we concluded that flower opening in P. umbraticola is under the control of the circadian clock. This conclusion was solely based on physiological experiments, and the assumption that the clock in P. umbraticola should not drastically differ from that in A. thaliana. Generation of P. umbraticola ESTs would allow us to verify this conclusion. Therefore, we report here the first comprehensive gene expression information on P. umbraticola based on high-throughput RNA-sequencing, and its application to the discovery of transcripts associated with the plant circadian clock and the ethylene biosynthesis and signaling pathways. Moreover, we characterized P. umbraticola core clock components, verifying our previous conclusion, and simultaneously demonstrated the depth and usefulness of our established ESTs.
Materials and Methods
Plant material and RNA extraction for RNA-sequencing (RNA-seq)
Two P. umbraticola cultivars, ‘Single Red’ (SR) and ‘Sanchuraka Cherry Red’ (SCR) (purchased from Sakata Seed Corporation, Yokohama, Japan) were used in this study. The plants were raised in a plant growth chamber following conditions previously described (Maguvu et al., 2016). The plants were raised in a phytotron at 28/23°C day and night temperatures, respectively, under natural sunlight, in Tokyo Japan. The plants were grown in 15-cm plastic pots filled with a 3:1 mixture of granular soil and peat-based soil mix (Metro-Mix 360; Sun Gro Horticulture, Agawam, MA, USA). Before sampling, the plants were transferred to a growth chamber (Espec corp., Osaka, Japan), which was maintained at 25 ± 2°C with a 12 h photoperiod, and entrained for one week. Light was supplied by metal halide lamps (MT400DL/BUD; Iwasaki Electric Co., Ltd., Tokyo, Japan). The cultivar ‘SR’ was used for RNA-seq, and each tissue was harvested from at least four independent plants. The tissues were harvested from petals and leaves. Petal samples were collected at full opening stage which occurred at approximately Zeitgeber time (ZT) 3 and just before senescence (~ZT 12). Leaf samples were collected in the morning (ZT 0) and in the evening (ZT 12) (Fig. 1). Petal samples collected at full opening stage and just before senescence were designated as PA and PP, respectively. Leaf samples collected in the morning and evening were designated as LA and LP, respectively. Tissues were immediately frozen in liquid nitrogen and stored at −80°C until use. Total RNA was extracted by combining a modified CTAB method with LiCl precipitation, and subsequent purification by NucleoSpin RNA kit (Macherey-Nagel GmbH and Co. KG, Düren, Germany). The concentration and purity of extracted total RNA was estimated using a Q5000 spectrophotometer (TOMY Seiko Co., Ltd., Tokyo, Japan). A similar method was used for RNA extraction in all other experiments.
DNA library construction and HiSeq 4000 sequencing
Sequencing libraries were constructed using TruSeq RNA Sample Prep Kit v2 (Illumina Inc., San Diego, CA, USA). The extracted total RNA (~200 ng) was treated with DNase I Oligo (dT) beads to isolate mRNA, and the fragmented mRNA was used to construct cDNA. Quantification and qualification of the sample library was done using an Agilent 2100 Bioanalyzer and the ABI StepOnePlus Real-Time PCR System. Subsequently, the libraries were sequenced using Illumina HiSeq 4000.
Bioinformatics analysis
Raw reads were filtered for low quality, adaptor-polluted, and high content of unknown reads, using Illumina internal software. The quality of reads was checked using FastQC before further processing. Trinity (version: v2.0.6 parameters: --min_contig_length 150 --CPU 8 --min_kmer_cov 3 --min_glue 3 --bfly_opts '-V 5 --edge-thr=0.1 --stderr ') was used to perform de novo assembly. Thereafter, Tgicl (version: v2.0.6 parameters: -l 40 -c 10 -v 25 -O '-repeat_stringency 0.95 -minmatch 35 -minscore 35') was used to cluster transcripts into unigenes. The obtained unigenes were annotated against seven functional databases (NT, NR, GO, COG, KEGG, SwissProt, and InterPro). Segments of unigenes that best mapped to the functional databases in a priority order of NR, SwissProt, KEGG, and COG were selected as CDS, and displayed from 5′ to 3′ in FASTA format. Unigenes that could not be aligned to any database mentioned above were predicted by ESTScan (version 3.0.2).
Availability of data and materials
The raw sequencing data generated by Illumina HiSeq4000 were submitted to the DDBJ Sequence Read Archive (DRA; <https://www.ddbj.nig.ac.jp/dra/index.html>) with accession numbers (DRR19734–DRR19737).
Identification of transcripts associated with ethylene response and the circadian clock
Using the sequenced data, a local nucleotide database was created with the bioinformatics tool BioEdit. From this database, the blastn algorithm was used to search for ethylene biosynthesis, perception, and downstream signaling genes, as well as circadian clock-associated genes. A. thaliana gene sequences were used as queries. The lengths of the obtained sequences were determined, and multiple sequence alignments were produced using Clustal Omega. Phylogenetic analysis of the retrieved sequences, and those of A. thaliana, was done using MEGA7 (Kumar et al., 2016).
Real-time Q-PCR analysis of ethylene biosynthesis and ethylene perception genes during senescence of P. umbraticola cultivars
Stems of two P. umbraticola cultivars, SR (short flower longevity) and SCR (longer flower longevity) bearing flower buds were harvested a day before anthesis, and placed in test tubes containing distilled water. The test tubes were held in a plant growth chamber at 28/25°C day and night temperatures, respectively, with cool fluorescent lights (315 μmol·m−2·s−1). The lights were turned on at 7 a.m. and turned off at 7 p.m. Sampling was done at 2 h intervals starting 2 h after lights on (9 a.m.), when the flowers were almost fully open, and ending at 11 p.m. when the flowers were fully closed. To avoid wound-induced ethylene biosynthesis and signal transduction, the whole flower, including the calyx, was used for RNA extraction. One sample consisted of at least three different flowers. Total RNA was isolated as described above. cDNA was synthesized from 3 μg RNA using ReverTra Ace qPCR RT Master Mix (Toyobo Co., Ltd, Osaka, Japan) in a total volume of 10 μL. Gene-specific primers were designed using Primer3 Input V.0.4.0, and are listed in Table S1. P. umbraticola Ubiquitin 10 (PuUBQ10) was used as an internal control. For PCRs, 2 μL of cDNA was used as a template with 5 μL THUNDERBIRD SYBR qPCR Mix (Toyobo), as well as 0.6 μL of reverse and forward primers in a final volume of 10 μL. The following temperature parameters were used: pre-denaturation: 95°C for 60 s; denaturation: 95°C for 15 s; annealing: 65°C for 30 s; and extension: 72°C for 60 s, with a total of 40 cycles. This procedure was performed using Illumina Eco. PCR Ver. 2.0 (AS ONE Corporation, Osaka, Japan). All reactions were performed in at least two independent biological replicates.
Real-time Q-PCR analysis of the circadian clock associated components
P. umbraticola cultivar SR was transferred into a dark room (previously described by Maguvu et al., 2016), and the plants were allowed to acclimatize to 12 h light/12 h dark (12L/12D) cycles for at least three days before sampling started. Leaf samples were collected at 4 h intervals for a period of 24 h, starting at (ZT 0). Total RNA was isolated as described above. cDNA synthesis and PCR were performed as described for the ethylene-related genes. Table S1 shows the gene specific primers used. P. umbraticola Tubulin (PuTUB) was used as an internal control.
Results
RNA-sequencing and de novo assembly
We report the first comprehensive transcriptome information for P. umbraticola, which will be vital for future molecular genetic studies. The sequencing cDNA libraries were constructed using RNA extracted from P. umbraticola leaves collected in the morning (LA) and evening (LP), petals at full opening (PA), and just before senescence (PP), in order to maximize the diversity of transcripts expressed at different times, and in different tissues. After cleaning the raw reads, assembly, and clustering, the LA sample generated a total of 48,954 unigenes with a total length, average length, N50, and GC content of 42,636,289 bp, 870 bp, 1,414 bp, and 43.2%, respectively (Table 1). The LP sample generated 50,242 unigenes with a total length, average length, N50, and GC content of 43,114,194 bp, 858 bp, 1,390 bp, and 43.2%, respectively (Table 1). The PA and PP samples generated a total of 40,712 and 42,966 unigenes respectively, with total lengths, average lengths, N50, and GC contents of 36,410,005 bp, 894 bp, 1,489 bp, 43.19%; and 35,730,162 bp, 831 bp, 1,378 bp, and 43.46%, respectively (Table 1). Merging the unigenes from all the samples resulted in a total of 68,928 unigenes (Table 1). The total number of unigenes was greater than those of individual samples, demonstrating the importance of sampling different tissues, as well as sampling at different times of the day, to ensure transcript diversity. Although the cumulative total of unigenes from all the samples was much higher than the total unigenes, this reduction in total unigenes was expected, as ESTs have been shown to contain a high degree of redundancy. This redundancy was shown in most, if not all, known ESTs; for example, in apples, Malus domestica, and carnations (Newcomb et al., 2006; Tanase et al., 2012). The total length, average length, N50 and GC content of the total unigenes were 68,287,771 bp, 990 bp 1,621 bp, and 42.78%, respectively (Table 1).

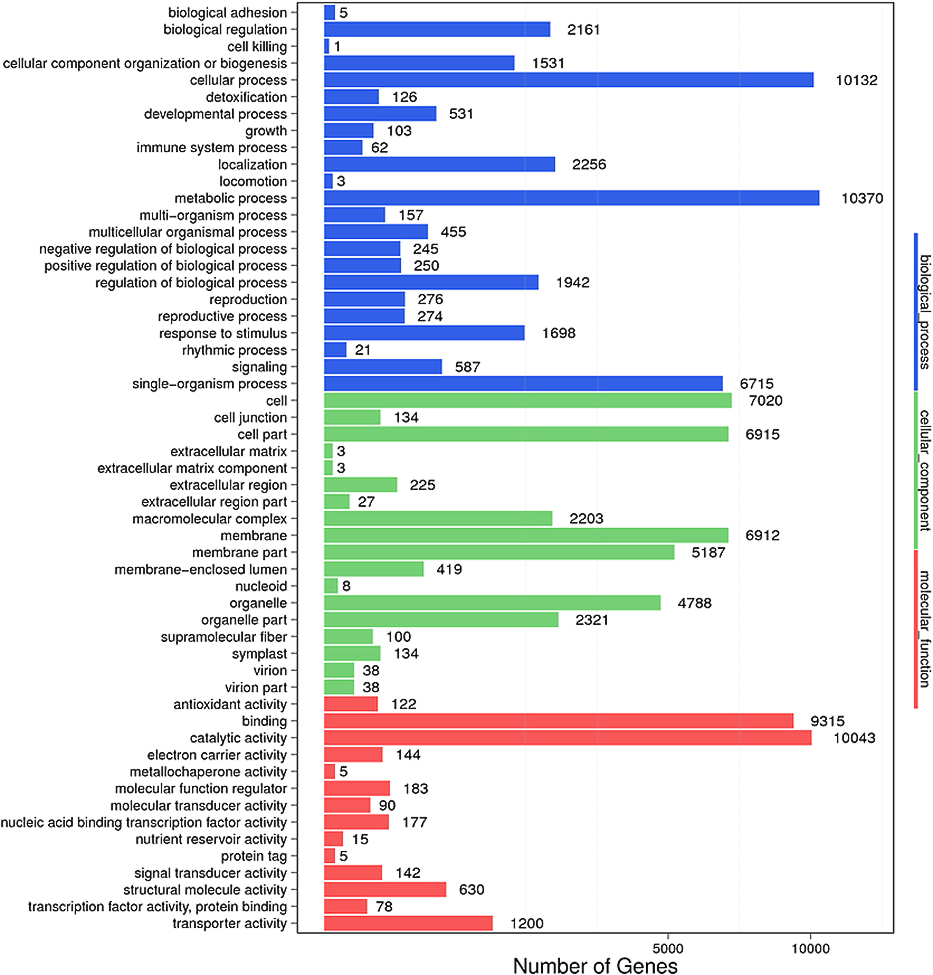
Unigene functional annotation was performed using seven functional databases, and the annotated results were as follows 40,995 (NR: 59.48%), 34,789 (NT: 50.47%), 28,250 (Swissprot: 40.98%), 16,948 (COG: 24.59%), 31,046 (KEGG: 45.04%), and 30,553 (Interpro: 44.33%) (Table 2). After functional annotation, the segment of the unigene that best mapped to functional databases was predicted as its CDS, for unannotated unigenes, ESTscan was used to predict the CDS (Table 3). The genes covered a broad range of Gene Ontology (GO) categories, with the main categories being biological processes, cellular components, and molecular functions. Distributions within each category are shown in Figure 2. The most common assignments in the biological processes category were metabolic processes (26%), cellular processes (25%), single-organism processes (17%), localization (6%), and biological regulation (5%). In the cellular components category, the largest classes were cell (19%), cell part (19%), membrane (19%), and organelle (13%). In the molecular function category, the largest classes were catalytic activity (45%), binding (42%), transporter activity (5%), and structural molecule activity (3%).

ESTs are a vital source of SSR, which are useful random markers for genetic population studies. Software MISA (version v1.0, parameters: 1-12,2-6,3-5,4-5,5-4,6-4 100 150 website: <http://pgrc.ipk-gatersleben.de/misa>) was used to screen SSR from the unigenes. A total of 4,657 (di), 4,538 (tri), 388 (quad), 611 (penta), and 1,335 (hexa), nucleotides were obtained (Fig. 3). The results indicate that 15.6% of the unigenes contained at least one SSR. In apples, carnations, and Cucurbita pepo, SSR motifs fall within a range of 3–20% (Blanca et al., 2011; Newcomb et al., 2006; Tanase et al., 2012). Although further studies are required to validate the usefulness of these markers, these data provide a platform and the tools required for identification of markers related to important traits.
Expression profiles of the transcripts in leaves and petals
The 12,000 most highly-expressed unigenes both in petal and leaf tissues are displayed by heatmap analysis (Fig. 4). We applied K-means clustering to interrogate tissue and sampling time-specific expression profiles of the transcripts. Figure 5 shows 20 clusters of transcripts with highly variable expression patterns among the samples. PP, PA, LA and LP had clusters of 1,370, 1,227, 512, and 711 highly expressed transcripts, respectively, as compared to all the other samples (cluster S and T, D, I, and G, Fig. 5). Tissue and sampling time-specific expression profiles of the transcripts were observed among the samples (Figs. 4 and 5).
Identification of the transcripts associated with ethylene biosynthesis, perception, and signaling pathways
Using the tblastn algorithm on a locally created nucleotide database, we were able to identify every transcript involved in the ethylene biosynthesis pathway (Table 4; Fig. S1). The amino acid sequences of A. thaliana ethylene biosynthesis genes were used as a query. We identified a total of five ACS and two ACO genes. However, phylogenetic analysis and sequence alignment revealed that only two (PuACS1 and 2) of the five ACS genes were authentic ACS genes involved in ethylene biosynthesis (Figs. S1 and S2). Two (PuACS3 and 4) of the five genes clustered with aminotransferases (AtACS10, AtACS12), which are not involved in ethylene biosynthesis, while another clustered with AtACS3, which is a known pseudogene. The pseudogene AtACS3 is a truncated version of AtACS1, which is enzymatically inactive as it is missing a highly conserved tripeptide (Liang et al., 1995), the P. umbraticola transcript (PuACS5) which clustered with AtACS3 is both truncated and lacks the highly conserved tripeptide (see conserved box 4 in Fig. S2), suggesting that it is also a pseudogene. The two authentic ACS genes were designated as PuACS1 and PuACS2, with 61.9 and 65.8% amino acid identity to AtACS6, respectively. The two ACO genes of P. umbraticola were 76.1 and 74.9% identical to AtACO4, and were designated as PuACO1 and PuACO2, respectively (Table 4).

We also identified ethylene receptors and downstream signaling genes. The query for the ethylene receptor genes produced a total of four hits, which were designated as PuETR1, PuETR2, PuERS1, and PuEIN4, respectively (Table 4). We also identified one REVERSION TO ETHYLENE SENSITIVITY (RTE) gene (PuRTE), three CTR1 (PuCTR1, 2, 3), and one EIN2 (PuEIN2) gene. Two genes encoding EIN3-like transcription factor (PuEIN3, PuEIL1), three EIN3-BINDING F-BOX protein (PuEBF1, 2, 3) and two ERF genes (PuERF 1, 2) were also identified (Table 4).
Characterization of the ethylene biosynthesis, and ethylene perception genes during senescence of P. umbraticola cultivars
P. umbraticola flowers were shown to have ethylene dependent senescence, with the cultivar SR showing a typical climacteric rise in ethylene production (Maguvu et al., 2016). Two cultivars (SR and SCR), which differ significantly in endogenous ethylene production and flower longevity, were used to characterize the ethylene biosynthesis and ethylene perception genes from flower opening until closure. Cultivar SR has shorter flower longevity and produces more endogenous ethylene than cultivar SCR (Maguvu et al., 2016). The transcripts of ethylene biosynthesis genes PuACS1 and PuACO2 were higher and peaked earlier in the SR cultivar than in the SCR cultivar (Fig. 6), which was consistent with observed endogenous ethylene production (Maguvu et al., 2016). At 14 h, in cultivar SCR, PuACS1 showed a higher transcript level than in cultivar SR, and at this time point, cultivar SR had already senesced. This was consistent with the FPKM value of RNA-seq data, which showed that the transcripts of PuACS1 and PuACO2 were upregulated in petals just before senescence (Table 4). In contrast to PuACS1 and PuACO2, PuACS2 and PuACO1 showed an expression pattern which was not consistent with ethylene production during senescence of P. umbraticola (Table 4, data not shown). Thus, PuACS2 and PuACO1 may be involved in system 1 ethylene regulation, which does not directly correlate with flower senescence as flower senescence is controlled by system 2 ethylene regulation (Barry et al., 2000; Lelièvre et al., 1997). The transcripts of ethylene receptor gene PuETR2 were appreciably upregulated towards flower senescence in both cultivars, but the transcript levels were always higher in SR cultivars than in SCR cultivars (Fig. 6). Upregulation of PuETR2 coincided with an upregulation of PuACS1 and PuACO2 (Fig. 6). Among the ethylene receptor genes, only PuETR2 showed a dramatic increase in the FPKM value just before senescence (Table 4). In addition, expression of PuERF1 and PuERF2 increased just before petal senescence (Table 4).

The time of flower opening in P. umbraticola is affected by photoperiod, and the endogenous circadian clock plays a pivotal role in this process (Maguvu et al., 2018). We managed to identify homologs of A. thaliana circadian clock component genes, including LHY, CIRCADIAN CLOCK-ASSOCIATED 1 (CCA1), TIMING OF CAB EXPRESSION 1 (TOC1, also known as PRR1), PSEUDO RESPONSE REGULATORs (PRR5/7/9), EARLY FLOWERING 3/4 (ELF3/4), GIGANTEA (GI), REVEILLE (RVE), LUX ARRYTHMO (LUX also known as PHYTOCLOCK 1 (PCL1)), ZTL, and FLAVIN-BINDING, KELCH REAPT, F-BOX 1 (FKF1) (Nohales and Kay, 2016). The identified putative P. umbraticola homologs of LHY/CCA1, PRR9/7/5, PRR1/TOC1, LUX/PCL1, and GI were designated as PuCCA1, PuLHY, PuPRR9/7/5, PuTOC1, PuLUX, and PuGI, respectively (Table 4). Moreover, we also identified transcripts linked to the oscillator through the input and output pathways. We identified two hits for ZTL, FKF1 and a hit each for ELF3 and ELF4. They were designated as PuZTLa/b, PuFKF1a/b, PuRVE4/8, PuELF3, and PuELF4, respectively (Table 4).
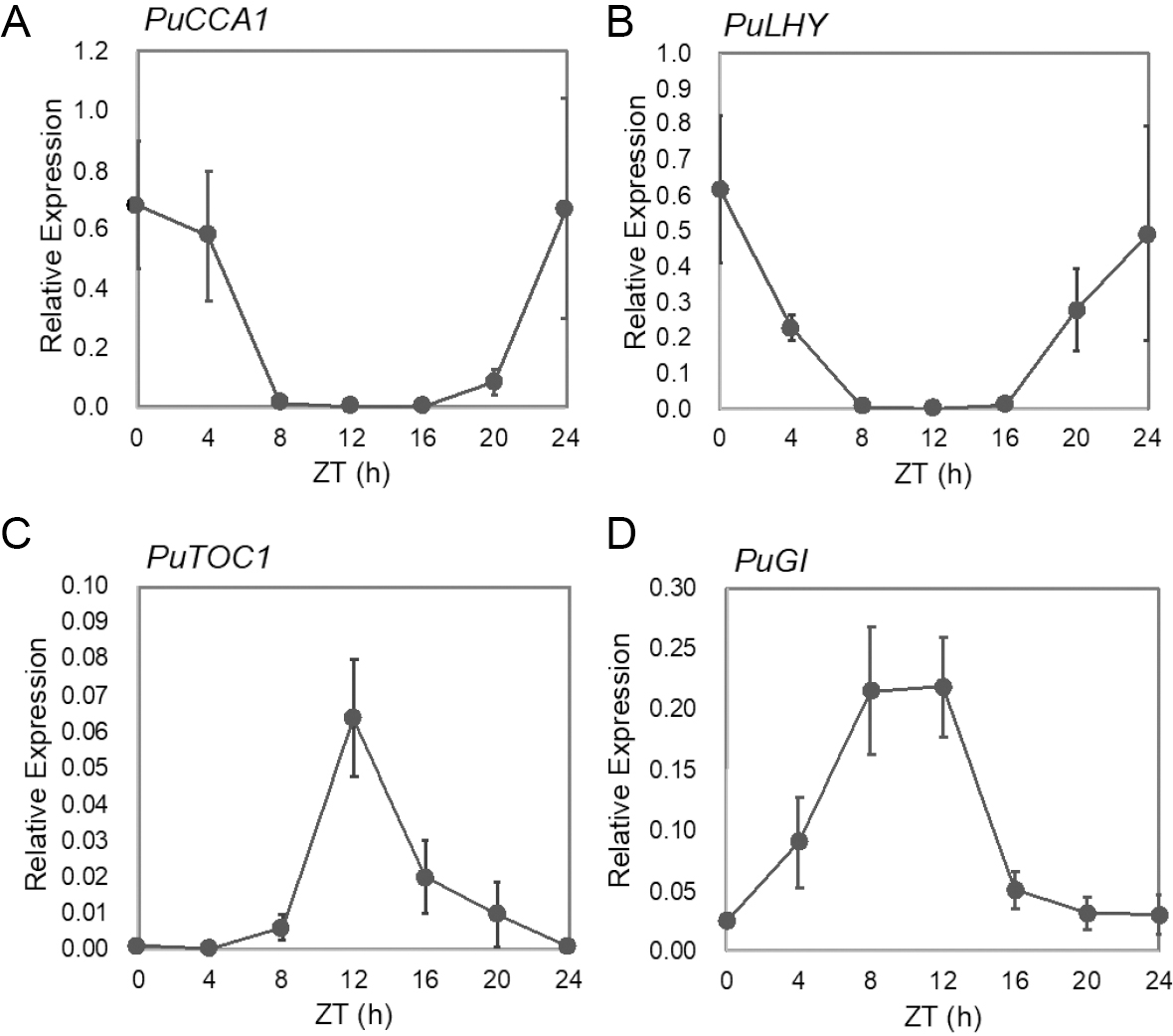
Most of the circadian clock component genes tested to date in many different plant species show robust diurnal rhythms under light/dark (L/D) cycles and free-running rhythms under constant conditions. The genes encoding the core clock components show diurnal rhythms that peak in the morning (LHY, CCA1), mid-day to afternoon (PRR5, PRR7), or evening (GI, TOC1) (Sanchez and Kay, 2016). Under a 12L/12D cycle, the transcripts of PuCCA1 showed a robust diurnal rhythm, peaking at dawn (ZT0), and then gradually decreasing, reaching their trough level at ZT8. This lower level was maintained up until ZT16, and from then onwards a gradual increase was observed, with transcripts of PuCCA1 peaking again at dawn (Fig. 7). The expression pattern of PuLHY mirrored that of PuCCA1 (Fig. 7). In contrast to PuLHY and PuCCA1, PuTOC1 transcripts were very low from ZT0 up to ZT4, and then from ZT8 the transcripts started to increase, and peaked at ZT12 (dusk). Thereafter PuLHY and PuCCA1, PuTOC1 transcripts showed a gradual decrease, reaching their trough levels at dawn (Fig. 7). Similarly, expression of PuGI showed a clear diurnal rhythm, peaking in the evening (Fig. 7).
Discussion
Expression profiles of the transcripts in leaves and petals
This report represents the first overview of P. umbraticola transcriptome profiling for leaves and petals using RNA-seq. To increase the diversity of transcripts, RNA was extracted from leaves in the morning and evening (LA and LP), as well as from petals at full opening (PA) and just before senescence (PP). We applied K-means clustering to interrogate tissue and sampling time specific expression profiles of the transcripts. Figure 5 shows 20 clusters of transcripts with highly variable expression patterns among the samples. For example, CL8678.Contig2_All (senescence-specific cysteine protease SAG12-like), which is a senescence-related gene was highly expressed in PP. SAG12 in A. thaliana is used as a standard marker for leaf senescence (Hensel et al., 1993; Lohman et al., 1994; Noh and Amasino, 1999). Moreover, the genes encoding cysteine proteases were reported to be upregulated during senescence in most flowers (Jones et al., 1995; Tripathi et al., 2009; Yamada et al., 2007). Thus, this observation raises interesting questions, as well as forming a basis for future research: How is this gene and the other genes highly expressed before senescence involved in the senescence process of P. umbraticola?; Can tinkering with any of these genes improve flower longevity? The ESTs presented in this study may pave the way for future studies to answer such questions.
On the other hand, most of the genes expressed in the leaves were housekeeping genes essential for plant survival; this is common in most if not all transcriptome analysis (Zhang et al., 2016), since leaves are the main source of photoassimilates. Moreover, tissue-specific expression patterns were also observed (Figs. 4 and 5). For example a number of genes encoding carbonic anhydrases (CAs) were only identified in leaf samples (Unigene5439_All, CL720.Contig5_All, CL8707.Contig1_All, and CL6859.Contig4_All). CAs are essential for photosynthetic organisms and their intracellular location is critical. αCA genes show quite specific organ or tissue expression patterns, and in most cases they are highly expressed in leaf tissue (Makita et al., 2015; Tang et al., 2014; Tanz et al., 2009). CAs were shown to impact stomatal movement and development (Hu et al., 2010), and limit the rate of photosynthesis (von Caemmerer et al., 2004). Thus, further analysis of their role in P. umbraticola may help understanding of heat stress tolerance and effective photosynthetic mechanism of this species. This knowledge could then help to improve less photosynthetically efficient crop plants.
Expressions of ethylene-related genes during flower senescence of P. umbraticola
A climacteric rise in ethylene production was shown to be accompanied by an increase in the expression of genes encoding ACS and ACO in most plants (Henskens et al., 1994; Tanase et al., 2008; Tang and Woodson, 1996; Wang and Woodson, 1991). In carnations, long-life cultivars produced little ethylene and had lower transcript levels of some ACS and ACO genes than short lived cultivars (Tanase et al., 2008). In this study, we found that PuACS1 and PuACO2 transcripts were correlated with endogenous ethylene production, as well as senescence of the two cultivars. Variation of flower longevity between cultivars of the same species can be due to low ethylene production, low ethylene sensitivity (Reid and Wu, 1992), or a combination of the two (Brandt and Woodson, 1992). In P. umbraticola, various doses of exogenous ethylene accelerated senescence of both SR and SCR cultivars (Maguvu et al., 2016), suggesting that both cultivars are sensitive to ethylene. Thus, the observed difference in flower longevity can be attributed to the difference in endogenous ethylene production, which correlates with the transcripts of PuACS1 and PuACO2, as well as senescence. PuACS2 and PuACO1 showed an expression pattern which did not correlate with endogenous ethylene production, suggesting that they may not play a role in senescence in P. umbraticola flowers. Ethylene receptors are known to be differentially regulated by ethylene, and the genes encoding these receptors show varying expression patterns during flower senescence in different species (Shibuya, 2012). In carnations, ERS2 and ETR1 did not show clear transcript changes. However, ERS1 decreased towards senescence (Shibuya et al., 2002). In A. thaliana, ETR1 and EIN4 are not appreciably regulated by ethylene, while the transcripts of ERS1, ERS2 and ETR2 are upregulated (Hua et al., 1998), in a manner similar to PuETR2. The upregulation is presumed to be a response mechanism to ethylene. Regulation of receptor genes probably serves to increase or decrease ethylene sensitivity by downregulating or upregulating expression of the receptors (Bleecker, 1999).
Conservation and diversification of circadian clock-related genes in P. umbraticola
The expression of circadian clock-component genes CCA1/LHY were shown to have a dawn phased peak (Boxall et al., 2005; Wang and Tobin, 1998). At their peak, CCA1 and LHY bind to the promoter of TOC1, thereby repressing its expression, resulting in the formation of a negative feed-back loop (Alabadi et al., 2000). Moreover, morning peaks of PuCCA1, PuLHY, and PuRVE4/8 were also confirmed by the FPKM value of the RNA-sequencing data. From the RNA-seq data, evening peaks of PuTOC1, PuPRR5a/b, PuGI, PuLUX, PuELF4, PuZTLb, and PuFKF1a were also observed (Table 4). The expression patterns of P. umbraticola core clock component genes were very similar to those observed in A. thaliana, suggesting that similar mechanisms may operate in these two species (Fig. 7). Somers et al. (2000) demonstrated that A. thaliana ZTL do not oscillate in continuous light or under LD cycles. However, our sequenced data suggest otherwise. Thus, we further characterized the PuZTLb transcripts under a 12L/12D cycle, and interestingly, these showed a robust diurnal rhythm, with a dusk-phased peak (Fig. S3A), suggesting a slight divergence from the A. thaliana clock. In the common ice plant, Mesembryanthemum crystallinum, which belongs to the order Caryophyllales and is known as a model of transition between C3 and CAM photosynthesis, ZTL homolog (McZTL) transcripts showed diurnal and circadian oscillation under LD and LL, respectively (Boxall et al., 2005). It is assumed that this may be related to the plant’s adaptations to extreme conditions. Phylogenetic analysis revealed that PuZTLb is closer to McZTL than AtZTL (Fig. S3B), suggesting that they may have evolved in a similar manner. Functional validation of PuZTLb and McZTL is essential to understand their roles.
Conclusions
This report presents the first significant comprehensive EST information for P. umbraticola, an invaluable new tool for biological research as it will enable characterization of the P. umbraticola transcriptome. We managed to isolate almost every gene involved in the ethylene biosynthesis and signaling pathways, as well as most core clock components, illustrating how useful our ESTs can be. Moreover, many other transcripts involved in other processes not in our area of interest can be also mined from this ESTs collection. Thus, we have paved the way for future molecular genetic studies on P. umbraticola. In addition, these ESTs helped us confirm that the operation of the plant circadian clock is largely conserved in P. umbraticola, although we did find some slight divergence.
Literature Cited
- Adams, M. D., M. B. Soares, A. R. Kerlavage, C. Fields and J. C. Venter. 1993. Rapid cDNA sequencing (expressed sequence tags) from a directionally cloned human infant brain cDNA library. Nat. Genet. 4: 373–380.
- Alabadi, D., T. Oyama, M. J. Yanovsky, F. G. Harmon, P. Mas and S. A. Kay. 2000. Reciprocal regulation between TOC1 and LHY/CCA1 within the Arabidopsis circadian clock. Science 293: 880–883.
- Barry, C. S., M. Immaculada Llop-Tou and D. Grierson. 2000. The regulation of 1-aminocyclopropane-1-carboxylic acid synthase gene expression during the transition from system-1 to system-2 ethylene synthesis in tomato. Plant Physiol. 123: 979–986.
- Blanca, J., J. Canizares, C. Roig, P. Ziarsolo, F. Nuez and B. Pico. 2011. Transcriptome characterization and high throughput SSRs and SNPs discovery in Cucurbita pepo (Cucurbitaceae). BMC Genomics 12: e104–118.
- Bleecker, A. B. 1999. Ethylene perception and signalling: an evolutionary perspective. Trends Plant Sci. 4: 269–274.
- Boxall, S. F., J. M. Foster, H. J. Bohnert, J. C. Cushman, H. G. Nimmo and J. Hartwell. 2005. Conservation and divergence of circadian clock operation in stress-inducible crassulacean acid metabolism species reveals clock compensation against stress. Plant Physiol. 137: 969–982.
- Brandt, A. S. and W. R. Woodson. 1992. Variation in flower senescence and ethylene biosynthesis among carnations. HortScience 27: 1100–1102.
- Gut, I. G. 2001. Automation in genotyping of single nucleotide polymorphism. Hum. Mutat. 17: 475–492.
- Hensel, L. L., V. Grbic, D. A. Baumgarten and A. B. Bleecker. 1993. Developmental and age-related processes that influence the longevity and senescence of photosynthetic tissues in Arabidopsis. Plant Cell 5: 553–564.
- Henskens, J. A. M., G. J. A. Rouwendal, A. ten Have and E. Woltering. 1994. Molecular cloning of two different ACC synthase PCR fragments in carnation flowers and organ-specific expression of corresponding genes. Plant Mol. Biol. 26: 453–458.
- Hu, H., A. Boisson-Dernier, M. Israelsson-Nordström, M. Böhmer, S. Xue, A. Ries, J. Godoski, J. M. Kuhn and J. I. Schroeder. 2010. Carbonic anhydrases are upstream regulators of CO2-controlled stomatal movements in guard cells. Nat. Cell Biol. 12: 87–93.
- Hua, J., S. Sakai, S. Nourizadeh, Q. C. Chen, A. B. Bleecker, J. R. Ecker and E. M. Meyerowitz. 1998. EIN4 and ERS2 are members of the putative ethylene receptor gene family in Arabidopsis. Plant Cell 10: 1321–332.
- Ichimura, K. and K. Suto. 1998. Environmental factors controlling flower opening and closing in a Portulaca hybrid. Ann. Bot. 82: 67–70.
- Jones, M. L., P. B. Larsen and W. R. Woodson. 1995. Ethylene-regulated expression of a carnation cysteine proteinase during flower petal senescence. Plant Mol. Biol. 28: 505–512.
- Kende, H. 1989. Enzymes of ethylene biosynthesis. Plant Physiol. 91: 1–4.
- Kumar, S., G. Stecher and K. Tamura. 2016. MEGA 7. Molecular Evolutionary Genetic Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33: 1870–1874.
- Kumari, K., M. Muthamilarasan, G. Misra, S. Gupta, A. Subramanian, S. K. Parida, D. Chattopadhyay and M. Prasad. 2013. Development of eSSR-Markers in Setaria italica and their applicability in studying genetic diversity, cross-transferability and comparative mapping in millet and non-millet species. PLoS ONE 8: e67742. DOI: 10.1371/journal.pone.0067742.
- Lara, M. Y., M. F. Drincovich and C. S. Andreo. 2004. Induction of crassulacean acid-like metabolism in the C4 succulent plant, Portulaca oleracea L.: Study of enzymes involved carbon fixation and carbohydrate metabolism. Plant Cell Physiol. 45: 618–626.
- Lelièvre, J. M., A. Latché, B. Jone, M. Bouzayen and J. C. Pech. 1997. Ethylene and fruit ripening. Physiol. Plant. 101: 727–739.
- Liang, X., Y. Oono, N. F. Shen, C. Kohler, K. Li, P. A. Scolnick and A. Theologis. 1995. Characterization of two members (ACS1 and ACS3) of the 1-ammino-cyclopropane-1-carboxylate synthase gene family of Arabidopsis thaliana. Gene (Amst) 167: 17–24.
- Lohman, K. N., S. Gan, J. Amasino, R. M. Amasino and C. Manorama. 1994. Molecular analysis of natural leaf senescence in Arabidopsis thaliana. Physiol. Plant. 92: 322–328.
- Maguvu, T. E., Y. Higuchi and M. Shibata. 2018. Effect of different photoperiods on flower opening time in Portulaca umbraticola. Hort. J. 87: 124–131.
- Maguvu, T. E., H. Shimizu-Yumoto and M. Shibata. 2016. Difference in flower longevity and endogenous ethylene production of Portulaca umbraticola cultivars. Hort. J. 85: 70–75.
- Makita, Y., S. Shimada, M. Kawashima, T. Kondou-Kuriyama, T. Toyoda and M. Matsui. 2015. MOROKOSHI: transcriptome database in Sorghum bicolor. Plant Cell Physiol. 56: e6. DOI: 10.1093/pcp/pcu187.
- Matthews, J. F., D. W. Ketron and S. F. Zane. 1992. Portulaca umbraticola Kunth (Portulacaceae) in the United States. Castanea 57: 202–208.
- McCarthy, D. L., G. Capitani, L. Feng, M. G. Gruetter and J. F. Kirsch. 2001. Glutamate 47 in 1-aminocyclopropane-1-carboxylate synthase is a major specificity determinant. Biochemistry 40: 12276–12284.
- Morgante, M., M. Hanafey and W. Powell. 2002. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 30: 194–200.
- Newcomb, R. D., R. N. Crowhurst, A. P. Gleave, E. H. A. Rikkerink, A. C. Allan, L. L. Beuning, J. H. Bowen, E. Gera, K. R. Jamieson, B. J. Janssen, W. A. Laing, S. McArtney, B. Nain, G. S. Ross, K. C. Snowden, E. J. F. Souleyre, E. F. Walton and Y. Yauk. 2006. Analyses of expressed sequence tags from apple. Plant Physiol. 141: 147–166.
- Noh, Y. S. and R. M. Amasino. 1999. Regulation of developmental senescence is conserved between Arabidopsis and Brassica napus. Plant Mol. Biol. 41: 195–206.
- Nohales, M. A. and S. A. Kay. 2016. Molecular mechanisms at the core of the plant circadian oscillator. Nat. Struct. Mol. Biol. 23: 1061–1069.
- Ocampo, G. and J. T. Columbus. 2012. Molecular phylogenetics, historical biogeography, and chromosome number evolution of Portulaca (Portulacaceae). Mol. Phylogenet. Evol. 63: 97–112.
- Onozaki, T. 2018. Breeding of carnations (Dianthus caryophyllus L.) for long vase life. Breed. Sci. 68: 3–13.
- Park, K. Y., A. Drory and W. R. Woodson. 1992. Molecular cloning of an 1-aminocyclopropane-1-carboxylate synthase from senescing carnation flower petals. Plant Mol. Biol. 18: 377– 386.
- Rafalski, J. A. 2002. Novel genetic mapping tools in plants: SNPs and LD-based approaches. Plant Sci. 162: 329–333.
- Reid, M. S. and M. J. Wu. 1992. Ethylene and flower senescence. Plant Growth Regul. 11: 37–43.
- Rottmann, W. E., G. F. Peter, P. W. Oeller, J. A. Keller, N. F. Shen, B. P. Nagy, L. P. Taylor, A. D. Campbell and A. Theologis. 1991. 1-Aminocyclopropane-1-carboxylate synthase in tomato is encoded by a multigene family whose transcription is induced during fruit and floral senescence. J. Mol. Biol. 222: 937–961.
- Sanchez, S. E. and S. A. Kay. 2016. The plant circadian clock: From a simple time keeper to a complex developmental manager. Cold Spring Harb. Perspect. Biol. 8: a027748. DOI: 10.1101/cshperspect.a027748.
- Shibuya, K. 2012. Molecular mechanisms of petal senescence in ornamental plants. J. Japan. Soc. Hort. Sci. 81: 140–149.
- Shibuya, K., M. Nagata, N. Tanikawa, T. Yoshioka, T. Hashiba and S. Satoh. 2002. Comparison of mRNA levels of three ethylene receptors in senescing flowers of carnation (Dianthus caryophyllus L.). J. Exp. Bot. 53: 399–406.
- Shinozaki, Y., R. Tanaka, H. Ono, I. Ogiwara, M. Kanekatsu, W. G. van Doorn and T. Yamada. 2014. Length of the dark period affects flower opening and the expression of circadian-clock associated genes as well as xyloglucan endotransglucosylase/hydrolase genes in petals of morning glory (Ipomoea nil). Plant Cell Rep. 33: 1121–1131.
- Somers, D. E., T. F. Schultz, M. Milnamow and S. A. Kay. 2000. ZEITULUPE encodes a novel clock-associated PAS protein from Arabidopsis. Cell 101: 319–329.
- Sreenvasulu, N., P. B. Kavi Kishor, R. K. Varsheny and L. Altschmied. 2002. Mining functional information from cereal genomes—the utility of expressed sequence tags. Curr. Sci. 83: 965–973.
- Tanase, K., C. Nishitani, H. Hirakawa, S. Isobe, S. Tabata, A. Ohmiya and T. Onozaki. 2012. Transcriptome analysis of carnation (Dianthus caryophyllus L.) based on next-generation sequencing technology. BMC Genomics 13: 292.
- Tanase, K., T. Onozaki, S. Satoh, M. Shibata and K. Ichimura. 2008. Differential expression levels of ethylene biosynthetic pathway genes during senescence of long-lived carnation cultivars. Postharvest Biol. Technol. 47: 210–217.
- Tang, H., V. Krishnakumar, S. Bidwell, B. Rosen, A. Chan, S. Zhou, L. Gentzbittel, K. L. Childs, M. Yandell and H. Gundlach. 2014. An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genomics 15: 312.
- Tang, X. and W. R. Woodson. 1996. Temporal and spatial expression of 1-aminocyclopropane-1-carboxylate oxidase mRNA following pollination of immature and mature petunia flowers. Plant Physiol. 112: 503–511.
- Tanz, S. K., S. G. Tetu, N. G. F. Vella and M. Ludwig. 2009. Loss of the transit peptide and an increase in gene expression of an ancestral chloroplastic carbonic anhydrase were instrumental in the evolution of the cytosolic C4 carbonic anhydrase in Flaveria. Plant Physiol. 150: 1515–1529.
- Tripathi, S. K., A. P. Singh, P. Sane and P. Nath. 2009. Transcriptional activation of a 37 kDa ethylene responsive cysteine protease gene, RbCP1, is associated with protein degradation during petal abscission in rose. J. Exp. Bot. 60: 2035–2044.
- von Caemmerer, S., V. Quinn, N. C. Hancock, G. D. Price, R. T. Furbank and M. Ludwig. 2004. Carbonic anhydrase and C4 photosynthesis: a transgenic analysis. Plant Cell Environ. 27: 697–703.
- Voznesenskaya, E. V., N. K. Koteyeva, G. E. Edwards and G. Ocampo. 2010. Revealing diversity in structural and biochemical forms of C4 photosynthesis and a C3-C4 intermediate in genus Portulaca L. (Portulacaceae). J. Exp. Bot. 61: 3647–3662.
- Wang, H. and W. R. Woodson. 1991. A flower senescence-related mRNA from carnation shares sequence similarity with fruit ripening-regulated mRNAs involved in ethylene biosynthesis. Plant Physiol. 96: 1000–1001.
- Wang, Z. Y. and E. M. Tobin. 1998. Constitutive expression of the CIRCADIAN CLOCK ASSOCIATED 1 (CCA1) gene disrupts circadian rhythms and suppresses its own expression. Cell 93: 1207–1217.
- Yamada, T., K. Ichimura, M. Kanekatsu and W. G. van Doorn. 2007. Gene expression in opening and senescing petals of morning glory (Ipomoea nil) flowers. Plant Cell Rep. 26: 823–835.
- Yon, F., Y. Joo, L. C. Llorca, E. Rothe, I. T. Baldwin, and S. G. Kim. 2016. Silencing Nicotiana attenuata LHY and ZTL alters circadian rhythms in flowers. New Phytol. 209: 1058–1066.
- Zhang, L., Y. Long, C. Fu, J. Xiang, J. Gan, G. Wu, H. Jia, L. Yu and M. Li. 2016. Different gene expression patterns between leaves and flowers in Lonicera japonica revealed by transcriptome analysis. Front. Plant Sci. 7: 637. DOI: 10.3389/fpls.2016.00637.