Abstract
Epigenetics is defined as “the study of changes in gene function that are mitotically and/or meiotically heritable and do not involve a change in DNA sequence”. Epigenetic modifications include post-translational modifications of histones and DNA methylation. Changes in DNA methylation have been observed in response to environmental factors, with some epimutations being heritable across generations. Such epimutations may lead to alterations in plant traits and could be involved in natural selection or domestication. Furthermore, epigenetic transcriptional regulation serves as a crucial strategy for responding to various environmental conditions, including abiotic and biotic stress. In horticultural crops, this regulation is implicated in diverse biological processes, including agronomic traits such as hybrid vigor/heterosis, flowering time, bud dormancy, sex determination, fruit ripening, and anthocyanin accumulation. Recent advances in methods for analyzing epigenetic states, along with advances in sequencing technology, have enabled high-resolution and genome-wide studies in various horticultural crops. This review highlights the critical role of epigenetic transcriptional regulation in biological processes in horticultural crops and discusses the potential for artificially inducing epigenetic variation to enhance phenotypic diversity in horticultural crop breeding.
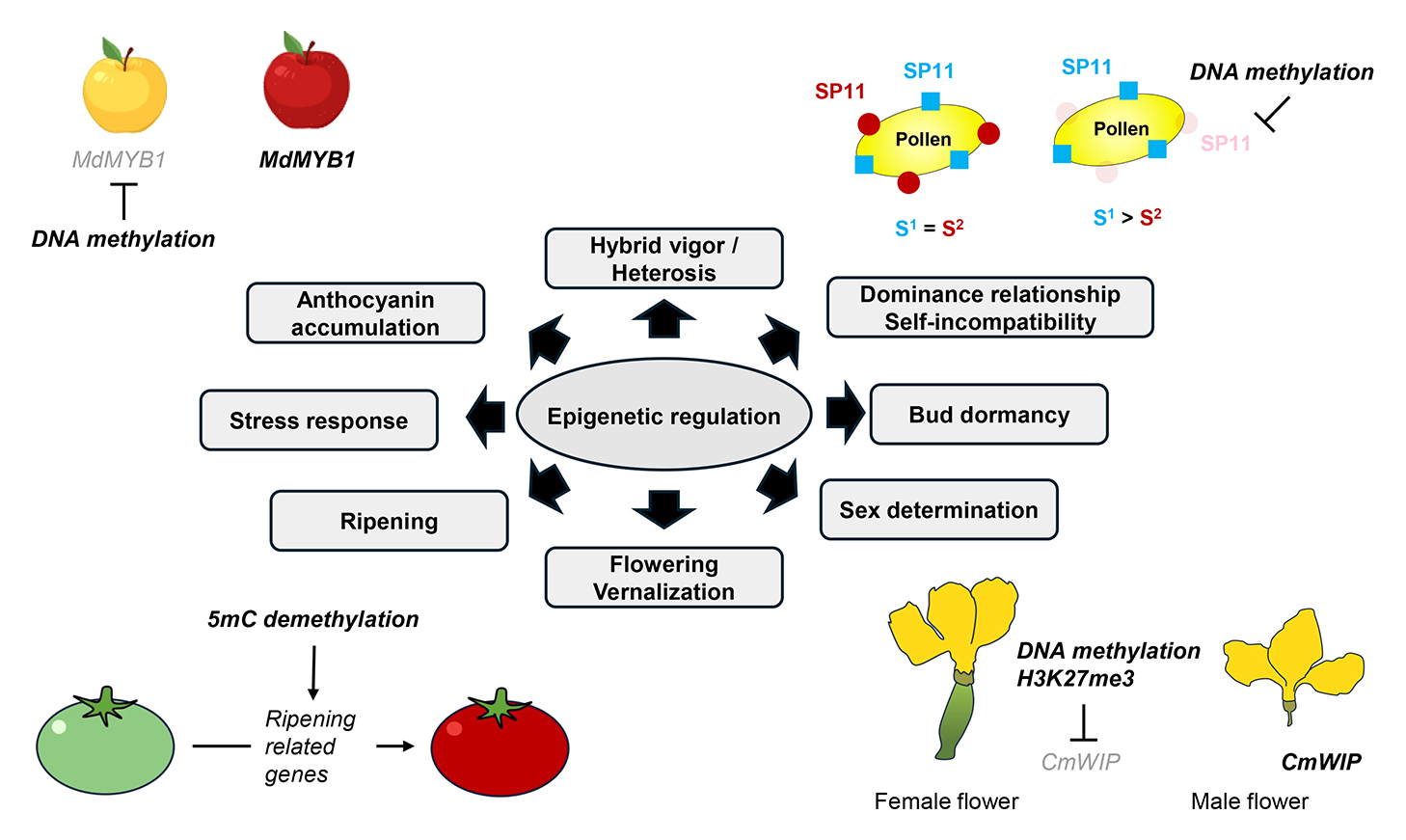
Introduction
The blueprint of an organism is specified by the order of its DNA bases, and DNA sequences define gene expression and protein function. Changes in DNA sequence in the genic regions can lead to changes in their expression, but gene expression can also be altered without any changes in the DNA sequence. The latter mechanism is called epigenetics. The word “epi” is a Greek prefix meaning “after” or “over” and was proposed in the 1940s as a field of study related to the regulatory mechanisms of gene expression that do not involve changes in DNA sequences. Originally describing a discipline, the phenomenon itself was later included in the meaning of epigenetics. Different kinds of epigenetic modifications, such as DNA methylation, chromatin remodeling, and histone posttranslational modification, can positively or negatively regulate gene expression (Fujimoto et al., 2012a; Kawakatsu and Ecker, 2019). These epigenetic modifications are involved in various biological processes that can regulate the transcription of genes related to agronomic traits in horticultural plants.
DNA methylation is a covalent addition of a methyl group (-CH3) to the fifth carbon position of cytosine (5mC). In plants, all sequence contexts of cytosines such as symmetric CG and CHG and asymmetric CHH (where H is A, C, or T) sites, are methylated. DNA methylation is preferentially observed in heterochromatic regions such as centromeric and pericentromeric regions, where many repetitive sequences and transposable elements (TEs) are included (Fujimoto et al., 2012a). DNA methylation in genic regions tends to be associated with transcriptional repression, while gene-body methylation (gbM), where only CG methylation is found in exon regions, is not associated with transcriptional repression (Fujimoto et al., 2012a). DNA methylation can be classified as maintenance DNA methylation and de novo DNA methylation. In the model plant Arabidopsis thaliana, DNA (cytosine-5) methyltransferase, METHYLTRANSFERASE 1 (MET1), CHROMOMETHYLASE 3 (CMT3), and CMT2 maintain the CG, CHG, and CHH methylation, respectively. RNA-directed DNA methylation (RdDM) is a biological process where noncoding RNA molecules direct the addition of DNA methylation to specific DNA sequences (Matzke and Mosher, 2014). RdDM is initiated when the precursor double-stranded RNA (dsRNA) is cleaved into 24 nucleotide small-interfering RNAs (24 nt-siRNAs) by the RNase III-like enzyme Dicer-like3 (DCL3). The RNA transcribed by DNA-dependent RNA polymerase V (Pol V) is used as a template for the silencing effector complex. The complementary siRNAs incorporated into the silencing effector complex form a base pair. The scaffold is thought to be the suppressor of TY insertion 5-like (SPT5l), which is an RNA-binding protein that can bind to Argonaute 4 (AGO4) and Pol V. Through this silencing effector complex, the de novo methyltransferase, named domains rearranged methyltransferase (DRM2), is recruited to the region of DNA corresponding to the siRNA and methylates the DNA. AGO4, DRM2, plus RNA-directed DNA methylation 1 (RDM1), which can bind to methylated DNA, may stabilize the site. DNA-dependent RNA polymerase IV (Pol IV) carries out transcription from the methylated DNA region. Pol IV transcripts are converted to dsRNA by RNA-dependent RNA polymerase 2 (RDR2), which is subsequently cleaved by DCL3, thereby amplifying the siRNA-induced DNA methylation system. In A. thaliana, DNA methylation is actively removed by the DNA glycosylase/lyase, DEMETER (DME), Repressor of silencing 1 (ROS1), and Demeter-like (DMLs) (Kawakatsu and Ecker, 2019; Zhang et al., 2018).
A nucleosome is the basic unit of eukaryotic chromatin, and this consists of 147 bp DNA wrapped around a core of histone proteins, the histone octamer (two each of the core histones H2A, H2B, H3, and H4). Modifications of the N-terminal tails of the core histones, such as methylation, acetylation, phosphorylation, and ubiquitination, regulate chromatin structure. The acetylation of lysine residues of histones H3 and H4 (i.e., K9, K14, K27ac) is reversible; adding an acetyl group to a lysine residue of histones is catalyzed by histone acetyltransferase (HAT) and removal of the acetyl group is catalyzed by histone deacetylase (HDAC). Histone lysine methylation is catalyzed by histone lysine methyltransferase (HKMTase) that contains SET [SU(VAR)3-9, enhancer of zeste E(z), and trithorax (TRX)] domains. Histone demethylases (HDMases) have two major domains, JUMONJI DOMAIN-CONTAINING PROTEIN C (JmjC) and lysine-specific demethylase domains (LSD) (Candela-Ferre et al., 2024). Lysine residues of core histones can be mono, di, or trimethylated, and each histone methylation has a different function in regulating gene expression (Fuchs et al., 2006; He et al., 2011). In plants, histone deacetylation and methylation such as H3K9me2 (dimethylation of lysine 9 of histone H3) and H3K27me3 are associated with gene repression, whereas histone acetylation and methylation such as H3K4me3 and H3K36me3 are associated with gene activation (Candela-Ferre et al., 2024).
The identification and biological significance of genes involved in epigenetic regulation is progressing in the model plant A. thaliana. The findings from A. thaliana can be generalized to plants at the basic level, but species-specific differences have been observed, and horticultural plants are no exception. Epigenetics research in horticulture has increased recently, and this review documents the progress of epigenetic research in horticultural plants.
Epigenome analysis
The epigenome is defined as a map of epigenetic marks such as DNA methylation or histone modification decorating the genome (Lloyd and Lister, 2022). The innovation of high-throughput sequencing technologies enables us to determine epigenome information in horticultural plants.
Methylome
Whole genome bisulfite sequencing (WGBS) is an approach to examine cytosine methylation at single-base resolution throughout the genome; genomic DNA is treated with sodium bisulfite (which converts unmethylated cytosine to uracil, but methylated cytosine is unchanged) before sequencing, and whole genome sequences are determined. The DNA methylation level at each cytosine can be calculated from the percentage of converted and unconverted cytosines. WGBS has been successfully applied to genome-wide DNA methylation profiling at single-base resolution in various plant species including horticultural plants (Niederhuth and Schmitz, 2014). However, bisulfite treatment can cause DNA degradation, so enzymatic methyl-seq (EM-seq) using the enzymatic conversion method was developed to study methylomes (Vaisvila et al., 2021). As an alternative to these cytosine conversion methods, Nanopore sequencing can identify DNA base modifications at single-nucleotide resolution, including 5mC, without any chemical treatments. Nanopore sequencing also has an advantage over WGBS in acquiring repetitive sequences data due to the long read length (Chen et al., 2022a).
Combining DNA methylation profile and transcriptome information can help to understand the relationship between DNA methylation states and transcript levels. In the case of Brassica vegetables, the average level of methylation of the whole genome was 36.5% in CG, 13.4% in CHG, and 5.3% in CHH sites using 14-day first and second leaves from an inbred line of Chinese cabbage (Brassica rapa) (Takahashi et al., 2018), 39.3% in CG, 15.4% in CHG, and 5.2% in CHH sites in the four-leaf stage of Chinese cabbage (Liu et al., 2018a), and 54.9% in CG, 9.4% in CHG, and 2.4% in CHH sites in Brassica oleracea (Parkin et al., 2014). Although the two groups used different stages and accessions in B. rapa, both showed that CG methylation in the promoter regions is associated with a decrease in gene expression, CHG and CHH methylation in genic regions are negatively associated with gene expression, and CG methylation in exon regions (gbM) is associated with higher expression levels as in other species (Liu et al., 2018a; Takahashi et al., 2018). In B. oleracea, there is a non-linear relationship between CG methylation levels in genic regions and expression levels; a higher level of expression is associated with lower CG methylation levels and genes having moderate levels of gene expression tend to have gbM (Parkin et al., 2014).
In tomato, WGBS was performed in four stages of fruit development and leaves, and the average level of methylation of the whole genome was 73.97–85.51% for CG, 51.99–56.15% for CHG, and 8.63–14.20% for CHH sites. CG and CHG methylation levels in leaves are higher than in fruit, while CHH methylation in fruit level is higher than in the leaves (Zhong et al., 2013). Around 60% of CG methylation is observed in gene-rich regions, while 85% of CG methylation is observed in gene-poor regions. CHG and CHH methylation levels in gene-rich regions are lower than in gene-poor regions (Gouil and Baulcombe, 2016).
In apple, the average level of methylation of the whole genome is ~53.6% for CG, ~37.7% for CHG, and ~8.5% for CHH sites (Xu et al., 2018), and 60.91–62.49% in CG, 43.60–45.44% in CHG, and 15.27–16.54% in CHH sites (Liu et al., 2022a). Genes having CG methylation in their bodies showed higher expression levels, and there was an inverse correlation between CHG methylation level and gene expression (Xu et al., 2018). There was a positive correlation between DNA methylation and TE density, while a negative correlation with gene number was observed (Liu et al., 2022a).
In the almond variety ‘Nonpareil’, the average levels of methylation of the whole genome in six tissue types (leaf, flower, exocarp, mesocarp, endocarp, and seed coat) were 45.2–56.4% for CG, 14.6–29.4% for CHG, and 1.3–4.7% for CHH sites. DNA methylation levels were the lowest and highest in leaf and flower tissues, respectively (Liu et al., 2022a).
Overall, the results on DNA methylation levels, the distribution of DNA methylation levels, and the relationship between DNA methylation and gene expression in horticultural plants is similar to other plant species.
Histone modification
Chromatin immunoprecipitation sequencing (ChIP-seq) is a method of ChIP of DNA followed by sequencing for genome-wide profiling of histone modifications where highly specific antibodies for histone modifications are used (Furey, 2012; Park, 2009). In Brassica vegetables, genome-wide histone profiling of active histone marks (H3K4me3 and H3K36me3) and repressive histone marks (H3K9me2 and H3K27me3) were examined using ChIP-seq in 14-day leaves from inbred lines of Chinese cabbage (Akter et al., 2019; Mehraj et al., 2021a; Takahashi et al., 2018). ChIP-seq using H3K27me3 antibodies was performed in primary leaves at 14 days after germination in the B. rapa variety ‘yellow sarson’ (var. trilocularis) (Payá-Milans et al., 2019). H3K4me3, H3K36me3, and H3K27me3 marks were found in euchromatic regions, while H3K9me2 was found in heterochromatic regions (Akter et al., 2019; Mehraj et al., 2021a; Payá-Milans et al., 2019; Takahashi et al., 2018). H3K4me3 and H3K36me3-marked genes were associated with gene activation (Mehraj et al., 2021a), while H3K9me2 and H3K27me3-marked genes were associated with gene repression (Akter et al., 2019; Payá-Milans et al., 2019; Takahashi et al., 2018). H3K36me3 and H3K27me3 antagonistically co-existed, while bivalent active H3K4me3 and repressive H3K27me3 histone modifications (simultaneous presence of H3K4me3 and H3K27me3 on the same nucleosomes) were identified in some genes of B. rapa (Mehraj et al., 2021a). Interestingly, biotic and abiotic stress-responsive genes tended to have bivalent active and repressive histone modifications in B. rapa, suggesting that these bivalent active and repressive histone modifications may be associated with higher transcriptional sensitivity to biotic or abiotic stress (Mehraj et al., 2021a). Histone modifications in the region encoding long noncoding RNAs (lncRNAs) were also examined in B. rapa (Akter et al., 2022; Mehraj et al., 2021b).
ChIP-seq using H3K9ac, H3K4me3, and H3K27me3 antibodies was performed in ovaries of emasculated flowers [0-day postanthesis (DPA)] and 4 DPA young fruit of tomato. ChIP-seq using H3K4me3, H3K27ac, H3K27me3, and H3K36me3 antibodies was performed in shoots and roots, six hours after nitrogen treatment in tomato. More than 53%, 54%, and 19% of genes had H3K9ac, H3K4me3, and H3K27me3 marks, respectively. H3K9ac and H3K4me3-marked genes showed a high level of gene expression, while H3K27me3-marked genes had low or no expression levels (Hu et al., 2021). H3K4me3 and H3K36me3 marks were observed in genic regions, while H3K27ac and H3K27me3 were observed in genic regions, upstream regions of the genic region, and intergenic regions. Differences in the level of histone modification changes were observed among four histone marks, with H3K27ac being the most variable, while H3K4me3 and H3K36me3 responded to nitrogen treatment. In addition, the genes altered in the same modification were different in shoots and roots (Julian et al., 2023).
A ChIP-seq protocol has been established in strawberry, pear, peach, and grape. These studies examined the association between expression levels of individual genes and histone modifications (Canton et al., 2022; Gao et al., 2021; Hermawaty et al., 2023; Huang et al., 2020a). ChIP-seq using H3K27me3 antibodies was performed in dormant and dormancy-related buds in peach. The distribution of H3K27me3 marks was similar between the two samples, and H3K27me3 was observed in euchromatic gene-rich regions (de la Fuente et al., 2015). In genic regions, enrichment of H3K27me3 marks was observed in the body and the transcription start site (TSS). ChIP-seq using H3K4me3 antibodies was performed in the three developmental stages of the floral buds (beginning of dormancy, end of dormancy, and ecodormancy) of sweet cherry, and 671 genes showed changes in H3K4me3 level, some of which showed a positive association between H3K4me3 levels and expression levels (Vimont et al., 2020).
The role of histone modifications in horticultural plants has been investigated in agriculturally important traits in horticultural plants, such as bud dormancy and fruit ripening. Integrated analysis combining ChIP-seq, RNA-seq, and methylome on a genome-wide basis has only just begun in horticultural plants.
Biological meaning of epigenetics as revealed by mutants
In A. thaliana, many genes involved in epigenetic modifications have been identified by mutant analyses and their role in epigenetic regulation of biological processes has also been shown. In horticultural plants, mutants have been created by a Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system and the role of epigenetic regulation in plant development has been clarified.
In tomato, there is one copy of NUCLEAR RNA POLYMERASE D1 (SlNRPD1) and one of NUCLEAR RNA POLYMERASE D1B (SlNRPE1), which are components of the largest subunit of Pol IV and Pol V, respectively. In A. thaliana, there is no obvious phenotypic change in atnrpd1 and atnrpe1 mutants, while slnrpd1 and slnrpe1 mutants in tomato showed abnormal leaves and sterility (Gouil and Baulcombe, 2016). Similarly, a developmental abnormality in seed size was observed in braA.nrpd1 and braA.nrpe1 mutants of B. rapa (Grover et al., 2018). As seed abortion occurs after fertilization, RdDM function is required in maternal sporophytic tissue in B. rapa (Grover et al., 2018). Reduction of 24nt-siRNAs was observed in slnrpd1 and braA.nrpd1 mutants, but not in slnrpe1 and braA.nrpe1 mutants (Gouil and Baulcombe, 2016; Grover et al., 2018). These results were similar in A. thaliana; Pol IV, but not Pol V, is involved in the biogenesis of 24nt-siRNAs. In tomato, cmt3a and kryptonite (kyp) mutants that result in a genome-wide reduction of CHG methylation also showed disrupted growth in both vegetative and reproductive tissues, although cmt3 and kyp mutants showed no noticeable phenotypic defects in A. thaliana (Wang and Baulcombe, 2020).
In tomato, there are four genes encoding DNA glycosylase/lyase, one of which, SlDML2, is highly induced during fruit ripening. Knockout mutants of sldml2 produced by CRISPR/Cas9 showed inhibition of fruit ripening and this phenotype was similar to the knock-down mutants produced by RNA interference (RNAi) (Lang et al., 2017; Liu et al., 2015b). In the ros1 mutant of A. thaliana, the effect on changes in gene expression was limited, while altered expression was observed in thousands of genes in sldml2 (Lang et al., 2017). In apple, MdROS1 expression was induced by low temperature, and knock-down and over-expression of MdROS1 were associated with low and high anthocyanin accumulation following low-temperature treatments, respectively. Low-temperature treatments showed a decrease in DNA methylation levels in the promoter regions of the anthocyanin regulatory or biosynthesis genes, suggesting the contribution of low temperature-induced MdROS1 (Yu et al., 2022).
DECREASE IN DNA METHYLATION 1 (DDM1) was identified as the gene responsible for the hypomethylated mutant in A. thaliana (Vongs et al., 1993). In A. thaliana ddm1 mutants, developmental defects were not obvious in the first generation, but appeared over succeeding generations (Kakutani et al., 1996; Kawanabe et al., 2016a). Some were due to ectopic expression caused by hypomethylation in their genes, as observed in the fwa mutant, while others were genetic mutations due to the disruption of gene function by insertion of transposons, which had been activated by hypomethylation (Fujimoto et al., 2008a, 2011, 2012a). In komatsuna and tomato, there are two copies of the DDM1 gene. Using RNAi, a knock-down ddm1 mutant was generated in komatsuna, which showed reduced DNA methylation in the regions covering centromeric repeats or TEs. Transcriptional reactivation of TEs was observed, but no obvious phenotypic change was observed (Fujimoto et al., 2008b). In tomato, ddm1 mutants were generated by the CSIPR/Cas9 technology. Single mutants, Slddm1a or Slddm1b, did not show any developmental defects, while the double mutant Slddm1a/Slddm1b showed pleiotropic vegetative and reproductive phenotypes. Plant size was small, less pollen was produced, and viable offspring were not produced from small parthenocarpic fruits (Corem et al., 2018). Comparable results were observed in ddm1 mutants in rice and maize (Long et al., 2021). The difference between komatsuna and tomato could be due to the difference in knock-down or knock-out mutations, and knock-out mutants in komatsuna are needed to confirm this possibility. In the Slddm1a/Slddm1b double mutant, extreme hypomethylation in CG and CHG sites was observed, but only moderate CHH hypomethylation was seen, even in TEs. A small number of TEs were transcriptionally reactivated in the Slddm1a/Slddm1b double mutant (Corem et al., 2018).
When genes involved in DNA methylation lose function, phenotypic changes or defects are more severe in horticultural plants than in A. thaliana. The average DNA methylation level is higher in horticultural plants than in A. thaliana; the role of DNA methylation is to regulate TEs and the number of TEs in A. thaliana is lower than in horticultural plants. TEs are highly DNA methylated; therefore, there is a positive association between the proportion of TEs in the genome and the global DNA methylation level and genome size. The difference in phenotypic impact between horticultural plants and A. thaliana may be related to the number of TEs. Therefore, regulation by DNA methylation is likely to be more important for normal development in horticultural plants than in A. thaliana.
Involvement of DNA methylation in vegetative growth vigor
Heterosis or hybrid vigor is a phenomenon where heterozygous F1 progeny show superiority compared with the homozygous parental lines and has been applied to F1 hybrid cultivation (Fujimoto et al., 2012b, 2018; Shiraki et al., 2023). The relevance of epigenetics in heterosis is discussed to explain the molecular mechanism of heterosis (Miyaji and Fujimoto, 2018).
WGBS was performed in heterotic F1 hybrids and their parental lines of pak choi, and DNA methylation levels in the F1 were higher than parental lines (Liu et al., 2020). Heterosis is also observed in broccoli (B. oleracea L. var. italic), especially in terms of curd weight. The transcriptome and methylome of 70-day-old curds of hybrids and their parental lines were analyzed (Li et al., 2018). Methylation-dependent restriction-site associated DNA (MethylRAD) methylome analysis revealed slightly higher and additive DNA methylation states in F1 (Li et al., 2018).
Mutants of genes involved in DNA methylation such as ddm1, met1, and mutS homolog 1 (msh1) in A. thaliana were tested for their effects on heterosis. Hybrids between two lines with the same genetic background, but different DNA methylation states, showed growth vigor (Miyaji and Fujimoto, 2018; Shiraki et al., 2023). MSH1-induced heritable enhanced growth vigor has been observed in A. thaliana, as well as in other crops such as sorghum and tomato. Knock-down of MSH1 by RNAi showed phenotypic alteration in vegetative and reproductive tissues, and these altered phenotypes were inherited even though the transgene had been segregated out. A hybrid (termed epiF1) between offspring of MSH1-RNAi plants without the transgene and wild type was produced, and the growth of this F1 was at a similar level to the wild type. However, the F2 progeny (epiF2) showed growth vigor, and superior growth, i.e., total fruit yield and fruit number, and some epiF2 was inherited by the next generation (epiF3 and epiF4) (Yang et al., 2015).
Differences in DNA methylation levels between a heterotic F1 and its parental lines were observed, but most regions in F1 showed additive DNA methylation states. How the differences in DNA methylation states between F1 and parental lines relate to heterosis is unclear. It is not known whether the mechanism of growth vigor seen in hybrids between two lines with the same genetic background, but different epigenetic modifications or in subsequent generations, is the same as the heterosis seen in hybrids between two different accessions. However, epigenetic regulation may play a significant role in the expression of heterosis, since the level of heterosis was reduced in F1s that had lost the function of DDM1 in A. thaliana (Kawanabe et al., 2016a; Zhang et al., 2016).
Epigenetic regulation of vernalization-induced flowering and bud dormancy
Brassica leafy or root vegetables such as cabbage, Chinese cabbage, turnip, komatsuna, and pak choi require vernalization, which is the induction of flowering by exposure to the prolonged cold of winter. Premature bolting results in reduced quality and yield; therefore, high bolting resistance is a desirable trait for these vegetables (Takada et al., 2019). FLOWERING LOCUS C (FLC) is a key gene in vernalization, and histone modifications are involved in the regulation of FLC expression in vernalization (Akter et al., 2021). In B. rapa, before a prolonged cold period, the expressed FLC had active histone modifications such as H3K4me3 and H3K36me3 in the gene body (Mehraj et al., 2021a). During cold temperature conditions, the accumulation of H3K27me3 around the TSS of FLC, and FLC expression was repressed (Akter et al., 2019, 2020). When plants return to warm conditions, H3K27me3 accumulation spreads across the FLC genes maintaining stable silencing (Akter et al., 2019; Kawanabe et al., 2016b). In A. thaliana, the plant homeodomain-PRC2 (PHD-PRC2) complex represses FLC expression through the accumulation of H3K27me3 during prolonged cold conditions (Berry and Dean, 2015; Whittaker and Dean, 2017). The loss of function of the histone methyltransferase, CURLY LEAF (CLF), which has an E(z) domain and is a component of PRC2, showed early bolting in B. rapa (Huang et al., 2020b; Payá-Milans et al., 2019; Tan et al., 2021). In the brclf mutant, expression levels of BrFT and BrFLCs increased in Chinese cabbage, suggesting that increased BrFT activity may mask the effects of the upregulated BrFLC expression (Huang et al., 2020b). The loss of function of histone methyltransferase, SET DOMAIN GROUP 8 (SDG8), in B. rapa results in early bolting and downregulation of BrFLCs, suggesting that BrSDG8 is involved in the epigenetic regulation of BrFLCs; possibly activating BrFLCs by adding H3K36me2/H3K36me3 (Fu et al., 2020). In A. thaliana, three cold-induced lncRNAs, COOLAIR, COLD ASSISTED INTRONIC NONCODING RNA (COLDAIR), and COLDWRAP, are involved in silencing and maintaining stable repression of FLC. However, in B. rapa, only COOLAIR-like transcripts have been identified in one of the four BrFLC paralogs, and the COLDAIR-like transcript was not detected from any of the four BrFLC loci (Li et al., 2016; Shea et al., 2019). As H3K27me3 accumulation was confirmed in all four BrFLC paralogs (Akter et al., 2019), but how PRC2 is recruited to the BrFLC loci without lncRNAs during vernalization needs to be clarified.
In seasonal perennial fruit trees, bud dormancy in the winter season is important for ensuring flowering at the right time, and release from dormancy requires a certain level of low temperature. Dormancy is classified into three categories, endodormancy, paradormancy, and ecodormancy. The MADS-box transcription factor, DORMANCY-ASSOCIATED MADS-box (DAM), is a key player in bud dormancy. The expression level of this gene decreased during the winter seasons (prolonged cold) and showed a similar expression pattern to FLC in Brassicaceae. The relationship between the behavior of expression patterns and histone modifications was also examined in Rosaceae fruits. In peach, PpeDAM1, PpeDAM4, PpeDAM5, and PpeDAM6 had lower expression levels and higher levels of H3K27me3 in dormancy-released buds compared with dormant buds (de la Fuente et al., 2015). Both PpeDAM5 and PpeDAM6 expression levels decreased following cold treatments, and a repressed state was maintained after transfer to warm conditions. H3K27me3 accumulated in PpeDAM6 following cold treatments, which was maintained after transfer to warm conditions, while accumulation of H3K27me3 was obvious only after transfer to warm conditions (Zhu et al., 2020a). After cold treatment, H3K27me3 accumulated only around the TSS of PpeDAM4, and expression was downregulated (de la Fuente et al., 2015, Zhu et al., 2020a). PavDAM5 was highly expressed with elevated level of H3K4me3 around the TSS in December (at the end of endodormancy), and the expression levels and H3K4me3 levels were reduced in January (ecodormancy) in sweet cherry (Vimont et al., 2020). In apple, all four DAM genes were downregulated following 60 days of cold treatments and showed decreased H3K4me3 levels. There were increased H3K27me3 levels following 60 days of cold treatments in DAM1 around the TSS and DAM4 in the entire gene body.
A gene that shows homology to FLC, also a MADS-box transcription factor, may also be involved in bud dormancy. Of the four FLC genes, FLC1, FLC2, and FLC4 were upregulated following 60 days of cold treatment, while FLC3 was downregulated. FLC2 expression levels showed a positive and negative association with H3K4me3 and H3K27me3 levels, respectively (Chen et al., 2022b). In kiwifruit (Actinidia chinensis), expression of the FLC-like gene (AcFLCL) continued to increase during a low-temperature period. Thirty-day cold treatment induced AcFLCL expression and accumulation of H3K4me3 marks around TSS compared with before treatment, but no change in H3K27me3 was detected. Overexpression of AcFLCL led to earlier bud break, and lines edited by CRISPR/Cas9 showed delayed bud break, suggesting AcFLCL plays a role in bud break (Voogd et al., 2022). In addition to the MADS-box gene, EARLY BUD-BREAK 3 (EBB3), which encodes an AP2/ERF domain-containing transcription factor, was involved in bud dormancy, and the expression of EBB3 before and after the cold period was negatively associated with changes in H3K27me3 in poplar (Azeez et al., 2021).
Changes in histone modifications such as H3K4me3 and H3K27me3 following prolonged cold treatment have been examined by ChIP-seq in studies of vernalization and bud dormancy (Akter et al., 2019; Chen et al., 2022b; Xi et al., 2020; Zhao et al., 2024). Although there is an issue regarding how to eliminate noise from the effect of differences in the developmental stages of the samples being analyzed, there is potential for identifying important genes involved in vernalization or bud dormancy by identifying genes that show changes in histone modifications.
Epigenetic regulation of sex determination
Sex determination in plants is a highly dynamic and complex process that involves multiple regulatory layers. In contrast to other eukaryotic organisms where sex determination is often dictated by fixed genetic factors, many plant species exhibit plasticity in their sex determination pathways. This flexibility is attributed to the involvement of epigenetic regulation, including DNA methylation, histone modifications, and small RNAs. These epigenetic factors modulate gene expression by altering chromatin structure and RNA stability, allowing plants to adjust their reproductive strategies in response to internal and external cues. The diversity in plant sex determination is evident in the wide range of sexual systems observed across plant species.
In dioecious plants, where individuals are distinctly male or female, epigenetic regulation is involved to establish and evolve sex chromosomes. In papaya (Carica papaya), heterochromatin structures exclusive to the male-specific region of the Y chromosome exhibit substantial divergence and significantly higher levels of DNA methylation compared to the corresponding regions on the X chromosome. These point to a pivotal role for DNA methylation and heterochromatin formation in the initial phases of sex chromosome evolution (Zhang et al., 2008). In Silene latifolia, the absence of certain retrotransposon families on the Y chromosome is hypothesized to be due to sex-specific DNA methylation and heterochromatin formation, which suppresses the activity of TEs (Cermak et al., 2008; Kubat et al., 2014; Rodríguez Lorenzo et al., 2018). This epigenetic silencing may play a crucial role in preventing genomic instability and preserving the functionality of the sex chromosomes. In addition, differential distribution of histone marks, such as H3K27me3 and H3K9me2, between X and Y chromosomes regulates Y-specific genes including APETALA3 Y-specific (SlAP3Y) and suggests a potential dosage compensation mechanism for regulation between different sex chromosomes similar to those found in mammals (Rodríguez Lorenzo et al., 2020). In garden asparagus (Asparagus officinalis), higher CG and CHG methylation levels on the Y chromosome compared to the X chromosome suggest a role for DNA methylation in Y chromosome evolution (Li et al., 2021).
A complex interplay between DNA methylation and small RNA-mediated regulation controls sex-specific traits in dioecious plants. In the diploid persimmon, Diospyros lotus, sex determination is regulated by a Y-chromosome-encoded small RNA gene known as Oppressor of MeGI (OGI) mediates repression of the female-promoting gene encoded homeodomain transcription factors, Male Growth Inhibitor (MeGI) (Akagi et al., 2014). OGI-derived small RNAs initiate RNAi mechanisms and potentially induce DNA methylation of the MeGI promoter, leading to its silencing and promoting male development. In polyploid persimmon, insertion of the SINE element Kali within the OGI promoter, along with the resulting near-silencing of OGI expression, facilitates a more dynamic and reversible regulation of MeGI (Akagi et al., 2016). This flexible control mechanism allows genetically male trees to produce both male and female flowers. This reversible epigenetic regulation demonstrates how ploidy and epigenetic modifications can interact to modulate sexual identity in polyploid plants. Additionally, in poplar (Populus balsamifera), small RNA-mediated regulation and DNA methylation at an ortholog of the Arabidopsis Response Regulator 17 (ARR17) locus control its sex-specific expression, demonstrating the role of RdDM in stabilizing male and female identities (Bräutigam et al., 2017). In garden asparagus, DNA methylation regulates the expression of genes involved in the suppression of female traits and the promotion of male organ development (Li et al., 2021).
In monoecious plants, where flowers comprise both male and female organs, such as cucumber (Cucumis sativus) and melon (Cucumis melo), unisexual flowers develop through a combination of epigenetic modifications and hormonal regulation (Fig. 1). In cucumber, the expression of genes including ethylene-related AMINOCYCLOPROPANE-1-CARBOXYLIC ACID OXIDASE 3 (CsACO3) and the ethylene response AP2/DREB-type transcription factor gene (RAP2.4), which regulate ethylene biosynthesis and female flower development, is modulated by temperature-dependent DNA demethylation and small RNA activity (Lai et al., 2017). Small RNAs guide DNA methylation changes in response to environmental cues, altering gene expression and influencing the balance between male and female flower formation. In C. melo, the 1-aminocyclopropane-1-carboxylic synthase (CmACS-7) gene suppresses stamen development in female flowers (Fig. 1). In male flowers, a zinc-finger transcription factor, wound inducible protein 1 (CmWIP1) represses CmACS-7, leading to carpel abortion and the formation of male flowers (Fig. 1). DNA methylation changes at a transposon inserted in the CmWIP1 promoter cause CmWIP1 silencing, resulting in a transition from male to female flowers (Martin et al., 2009). At the chromatin level, the regulation of sex-specific gene expression is further fine-tuned by histone modifications. The histone mark H3K27me3 is present not only on hormone-related and flower development genes, but also on CmWIP1, suggesting its role in controlling unisexual flower development (Latrasse et al., 2017). Additionally, the CmLHP1 protein modulates chromatin structure by interacting with both H3K27me3 and DNA methylation (Fig. 1), further regulating CmWIP1 expression (Rodriguez-Granados et al., 2022).
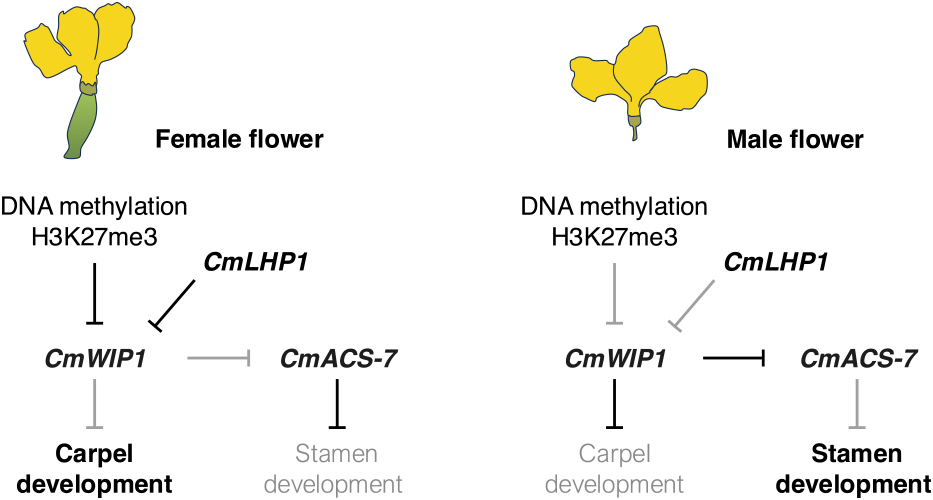
The interplay between DNA methylation, histone modifications, and small RNA-mediated regulation is central to the regulation of plant sex determination. These epigenetic mechanisms modulate sex-specific gene expression, TE activity, and chromatin state, contributing to the evolution of sex chromosomes and the establishment of sexual phenotypes in diverse plant species. Continued research in this field will enhance our understanding of plant reproductive biology and support the development of epigenetic-based breeding strategies for crop improvement.
Regulatory mechanism of the dominance relationship in the pollen determinant of self-incompatibility
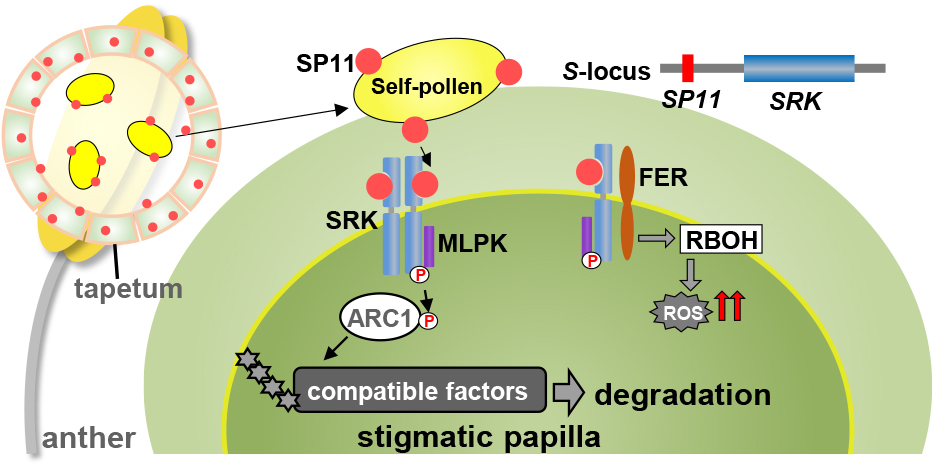
Many plant species have self-incompatibility (SI) mechanisms to prevent self-fertilization and promote outcrossing. This trait has been used for many years in the F1 hybrid breeding program of Brassica vegetables (Fujimoto et al., 2018). A single multi-allelic S-locus sporophytically controls the molecular mechanism of self/non-self-recognition of SI in Brassicaceae (Bateman, 1955). The S-locus of Brassicaceae contains two highly polymorphic genes; S-locus protein 11/S-locus cysteine-rich (SP11/SCR) encodes a small peptide containing conserved eight cysteine residues and is expressed in the anther tapetum cells and the S-locus receptor kinase (SRK) that encodes for a transmembrane-type receptor kinase. SRK is expressed in the stigma papillae cells (Schopfer et al., 1999; Suzuki et al., 1999; Takasaki et al., 2000; Takayama et al., 2000) (Fig. 2). S-alleles are called “S haplotypes” because these two genes are tightly linked and are inherited as one set. The S-haplotype-specific direct interaction of SP11 (pollen ligand) and SRK (stigma receptor) from the same S-haplotype plant induces the autophosphorylation of the SRK kinase domain and leads to self-pollen rejection on the stigma surface (Murase et al., 2020; Shimosato et al., 2007; Takayama et al., 2001). Several factors have been identified as downstream targets of SRK signaling. The plasma-membrane-anchored M-locus protein kinase (MLPK) is a positive regulator of SI reaction that is thought to interact with SRK (Murase et al., 2004). It is considered that the function of MLPK in SI was acquired in the genus Brassica (Ohata et al., 2023). Another SRK interactor is the arm-repeat-containing protein (ARC1) (Gu et al., 1998). ARC1 acts as an E3 ubiquitin ligase, which ubiquitinates the compatible pollination factors and degrades them by the proteasome (Indriolo et al., 2012; Stone et al., 1999) (Fig. 2). Recently, another receptor kinase, FERONIA (FER), has been reported to interact with SRK and has been suggested to induce the accumulation of reactive oxygen species (ROS) in the SI stigma. Downregulation of FER in the stigma resulted in a breakdown of the SI reaction (Abhinandan et al., 2022; Goring et al., 2023; Huang et al., 2023; Zhang et al., 2021) (Fig. 2).
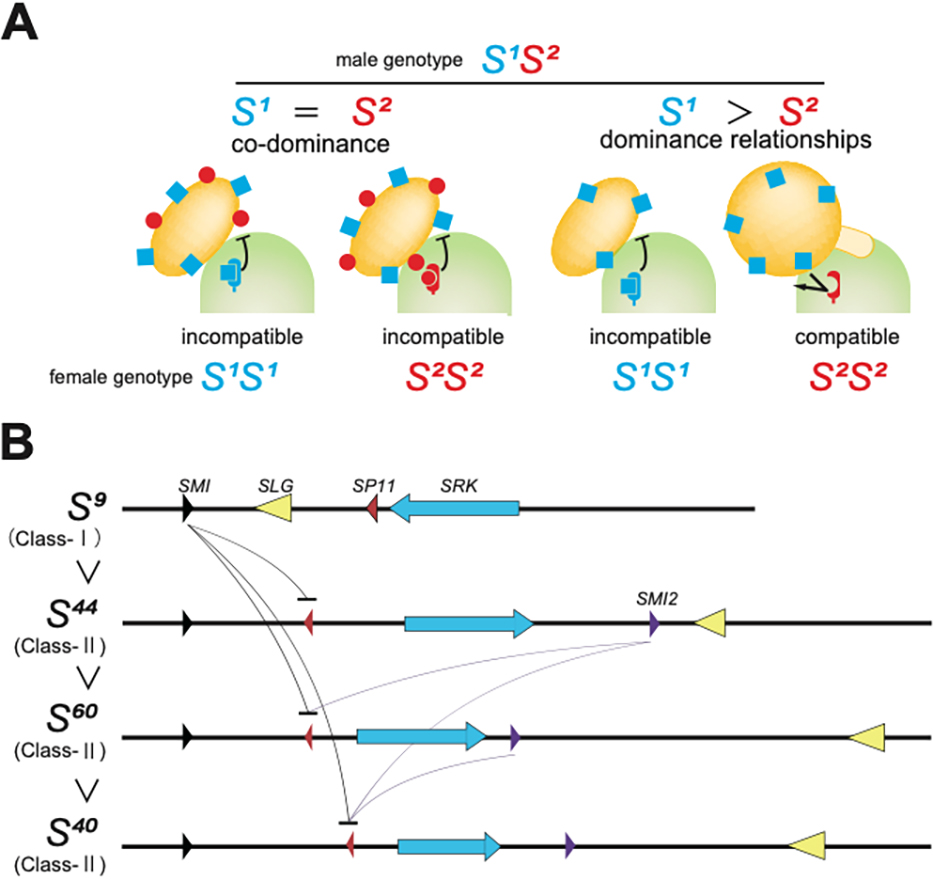
From the phylogenetic analysis of SRK, S haplotypes were classified into two classes (class I and class II) in Brassica. Over 100 S haplotypes have been estimated, and many S-haplotype lines have been isolated in B. rapa (Nou et al., 1993). Yamamoto et al. (2023) listed the S haplotypes and cognate sequences of SRK and SP11. Due to the nature of SI, there are many different S haplotypes within a given population, and each plant individual is likely to be heterozygous for the S haplotype. In the S-haplotype heterozygotes, individual haploid pollen grains exhibit S specificity (recognition identity of S haplotype) of both parental S haplotypes. However, for some heterozygous plants, the pollen was found to show an S-specificity for just one of the S haplotypes present in the parental genome (Fig. 3A). This relationship is known as the dominance relationship. In B. rapa, a detailed genetic analysis of the dominance relationship revealed the following three characteristics. (A) The class-I S haplotypes are generally codominant between the other class-I S haplotypes. (B) The class-I S haplotypes are dominant over class-II S haplotypes (class I > class II) (Hatakeyama et al., 1998; Thompson and Taylor, 1966). (C) Among the four class II S haplotypes (S29, S40, S44, S60 haplotype) in B. rapa, the dominant relationships are linear, S44 > S60 > S40 > S29, in which S44 is the most dominant and S29 is the weakest among four class-II S haplotypes (Kakizaki et al., 2003). How is the complex hierarchical structure of “many of class-I S haplotypes > S44 > S60 > S40 > S29” in the pollen-side dominant relationship controlled? The discovery of dominant S-haplotype-specific expression in the SP11 gene in the anther tapetum cells provided a breakthrough regarding this question. When the S haplotype of a heterozygous plant is codominant, the SP11 gene derived from both parental S haplotypes is expressed in anther tapetum cells, and the translated SP11 peptide is provided to the pollen grain carrying both S haplotypes (Shiba et al., 2002). However, in the heterozygous plants of S haplotypes with a dominant relationship, only expression of the SP11 gene from the dominant S haplotype in tapetum cells is detected, and the SP11 of the recessive S haplotype is suppressed (Shiba et al., 2002). The self-compatible B. rapa variety, ‘yellow sarson’ has a non-functional class-I S haplotype because it lacks SP11 gene expression. In heterozygotes derived from crossing cv. ‘yellow sarson’ and a homozygous class-II S haplotype line (SI), expression of SP11 in the allele of class-II S haplotype was suppressed (Fujimoto et al., 2006). This result indicates that a factor other than the expression of SP11 governs the suppression of class-II SP11 expression (Fig. 3B). Recessive SP11 gene expression was suppressed by de novo DNA methylation of the promoter region of the recessive SP11 allele. The DNA methylation of the SP11 promoter was controlled by an anther-specific trans-acting small noncoding RNA, named SP11 methylation inducer (Smi) (Tarutani et al., 2010). The sequences of Smi in class-I SP11s were similar to the target sequences of the recessive class-II SP11 promoter region (Tarutani et al., 2010). In the class-II S-haplotype, it was reported that another small noncoding RNA, which is called SP11 methylation inducer 2 (Smi2) that is produced at a different position to Smi induces de novo DNA methylation and suppresses the expression of recessive SP11 (Kakizaki et al., 2006; Yasuda et al., 2016) (Fig. 3B). An explanation for this is that hierarchical allelic repression based on sequence homology controls linear dominance relationships within the class-II S haplotypes. It has been suggested that the mechanism of small RNA-mediated epigenetic repression of SP11 is common throughout Brassicaceae (Fujii and Takayama, 2018). Further studies are needed to determine whether Smi, Smi2, near the S locus found in Brassica, is a common regulator of the dominance relationship in Brassicaceae. In a study of Arabidopsis halleri, at least 17 small RNAs were involved in the dominant relationship of SP11 expression, and it was recently reported that in Arabidopsis lyrata, three types of small RNAs produced by Smi1 and Smi2 regions like those in Brassica could explain the complex dominance relationship (Durand et al., 2014; Yasuda et al., 2021).
Epigenetic regulation of ripening
Fruit ripening is a biochemical process characterized by softening, color development, and enhanced flavor, which significantly influences the quality of horticultural crops. This process is regulated by plant hormone signals, including ethylene and a number of transcription factors (Liu et al., 2015a); studies on tomato have provided evidence that epigenetic mechanisms play a crucial role in the ripening process. The colorless non-ripening (cnr) mutant in tomato exhibits a phenotype of non-ripening of the fruits due to the silencing of the SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE-CNR (LeSPL-CNR) gene through hypermethylation in the promoter region (Manning et al., 2006; Zhong et al., 2013), demonstrating the impact of DNA methylation on ripening.
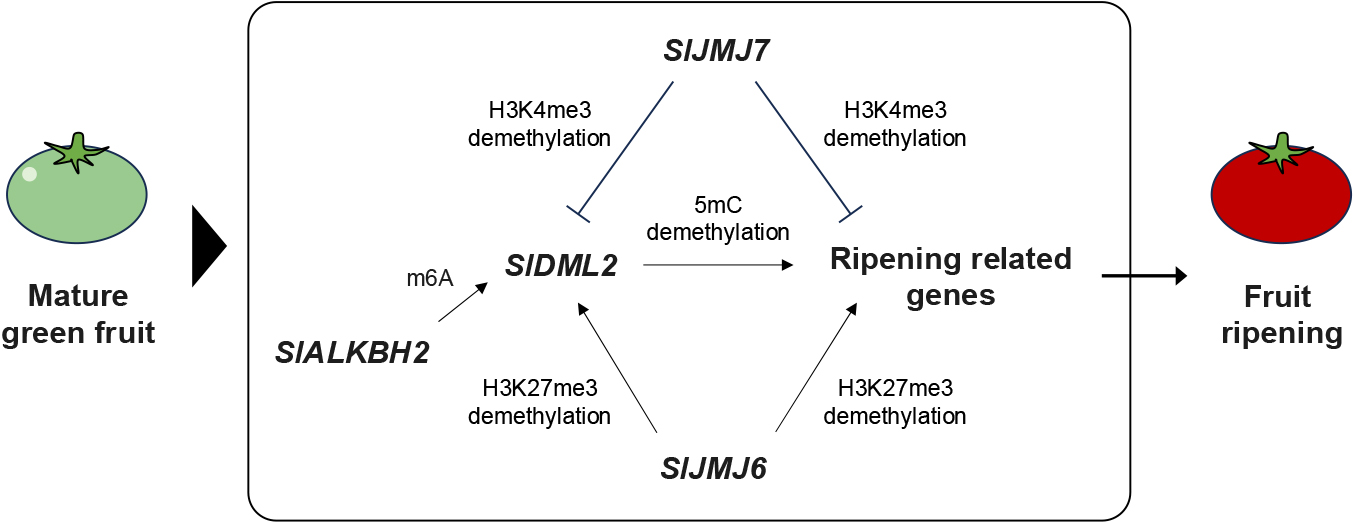
Genome-wide DNA methylation levels decrease significantly during tomato fruit development (Hu et al., 2021; Teyssier et al., 2008; Zhong et al., 2013). Treatment with the DNA methylation inhibitor 5-azacytidine accelerates the ripening process (Zhong et al., 2013), highlighting a significant relationship between DNA methylation changes and the fruit ripening process in tomato. A key factor in altering DNA methylation during ripening is the Solanum lycopersicum DEMETER-like DNA demethylase 2 (SlDML2) (Fig. 4). Knockout or knockdown lines of SlDML2 show a hypermethylation pattern and delayed ripening (Lang et al., 2017; Liu et al., 2015b). SlDML2-mediated demethylation activates ripening genes. Ripening genes such as NON RIPENING (NOR), PHYTOENE SYNTHASE 1 (PSY1), RIPENING INHIBITOR (RIN), and Vitamin E synthase 3 (VTE3) are naturally hypermethylated in their promoter regions in immature fruits and are demethylated in response to increased SlDML2 transcript levels during fruit development (Lang et al., 2017; Liu et al., 2015b). Hypermethylation in these promoters inhibits transcription factor binding, whereas demethylation by SlDML2 facilitates this binding, highlighting the critical role of SlDML2 in regulating fruit ripening (Liu et al., 2022b; Zhou et al., 2019). In addition, mRNA stability of SlDML2 is regulated through Solanum lycopersicum AlkB homolog 2 (SlALKBH2), which is involved in N6-methyladenosine (m6A) demethylation and stabilization (Zhou et al., 2019). Functional knockouts of SlALKBH2, similar to mutations in SlDML2, cause delays in fruit ripening, suggesting that RNA methylation indirectly regulates the ripening process via SlDML2.
Similar to tomato, the ripening process in strawberry is associated with changes in DNA methylation. DNA methylation levels decrease during ripening and methylation inhibitors can promote fruit ripening, highlighting the regulatory role of DNA methylation in strawberries (Cheng et al., 2018). In tomato, an increase in the DNA demethylase activity is a hallmark of the ripening process. Conversely, strawberries show no increase in DNA demethylase gene activity during ripening (Cheng et al., 2018). A downregulation of genes involved in the RdDM pathway, which uses small RNA molecules for transcriptional silencing, during ripening is accompanied by a decrease in the levels of siRNAs, suggesting reduced RdDM activity in the fruit ripening (Cheng et al., 2018). A recent study indicated that regulation of the furanone biosynthesis gene encoding quinone oxidoreductase (FaQR3), critical for flavor development during ripening, is controlled by DNA methylation and demethylation (Li et al., 2023). Knockdown lines of strawberry FaAGO4, a key component of the RdDM pathway, exhibit accelerated ripening and increased expression of FaQR3 due to increased DNA methylation levels in its promoter. Conversely, overexpression of FaAGO4 results in delayed ripening and decreased FaQR3 expression (Li et al., 2023). Knockdown lines of two strawberry demethylases FaDMLs delayed ripening and reduced FaQR3 expression (Li et al., 2023). These observations suggest a possible interplay between the RdDM pathway and DNA demethylation in the ripening process of strawberries. Sweet oranges show an increase in global DNA methylation levels during ripening, which is attributed to decreased expression of DNA demethylase genes, highlighting species-specific differences in the mechanisms by which DNA methylation influences ripening (Huang et al., 2019).
The ripening process in tomato is also influenced by histone modifications, particularly through the repressive H3K27me3 mark (Fig. 4), which has been shown to negatively correlate with the expression of ripening-related genes (Lü et al., 2018). A decrease in the expression of tomato Enhancer of Zeste (SlEZ2), a component of the PRC2 mediated H3K27me3 mark, leads to changes in fruit development and the ripening process (Boureau et al., 2016). Similarly, overexpression of another PRC2 component, MSI1, suppresses the transcription of ripening-related genes and results in delayed ripening (Liu et al., 2016). These results underscore the crucial role of transcriptional repression through PRC2-mediated H3K27me3 regulation in fruit development and the ripening process in tomato. Additionally, knockdown lines of a component of PRC1, tomato Like Heterochromatin Protein 1b (SlLHP1b), show significant upregulation of ripening-related genes including RIN and NOR, along with decreased H3K27me3 levels at these genes, thereby accelerating the ripening process (Liang et al., 2020). Conversely, overexpression of SlLHP1b led to down-regulation of ripening-related genes with enriched H3K27me3, which delayed ripening (Liang et al., 2020), suggesting that SlLHP1b regulates fruit ripening by epigenetically repressing ripening-related genes through PRC2-mediated H3K27me3.
Two tomato histone demethylases also play a role in regulating the expression of ripening-related genes. SlJMJ6 encodes a Jumonji C-terminal domain-containing histone lysine demethylase specifically for H3K27 methylation. Overexpression of SlJMJ6 accelerates tomato fruit ripening by upregulating the expression of ripening-related genes through decreased H3K27me3 levels (Li et al., 2020). In addition, SlJMJ6 directly activates DNA demethylase, SlDML2, by removing H3K27me3, implying that it could result in DNA demethylation and upregulated expression of ripening-related genes (Li et al., 2020). Another epigenetic histone mark, H3K4me3, which is associated with transcriptional activation, also plays a role in regulating tomato fruit ripening. Overexpression of SlJMJ7, an H3K4 demethylase, inhibits ripening, whereas knockdown lines of SlJMJ7 promote ripening (Ding et al., 2022). Although SlJMJ7 suppresses the ripening-related genes and DNA demethylase, SlDML2 in tomato fruit, expression of SlJMJ7 decreases during fruit development and leads to fruit ripening (Ding et al., 2022). Intriguingly, both SlJMJ6 and SlJMJ7 target the DNA demethylase enzyme DML2, suggesting that DML2’s activity in the ripening process is modulated positively and negatively by SlJMJ6 and SlJMJ7, respectively (Ding et al., 2022; Li et al., 2020). While many aspects of epigenetic control in the ripening process are still under investigation, these studies present new opportunities for enhancing fruit quality in horticultural crops through genetic and epigenetic interventions.
Epigenetic regulation of expression of genes involved in anthocyanin accumulation
Anthocyanins, a class of plant flavonoids, play a crucial role not only in human health owing to their antioxidant properties, but also in imparting color to fruit skin or flowers in ornamental plants. The biosynthesis of anthocyanins is regulated by structural genes and transcription factors that modulate the expression of these structural genes. DNA methylation has been shown to be involved in the transcriptional regulation of anthocyanin biosynthesis regulatory genes, with studies indicating an association between DNA methylation patterns and anthocyanin accumulation across various horticultural plants.
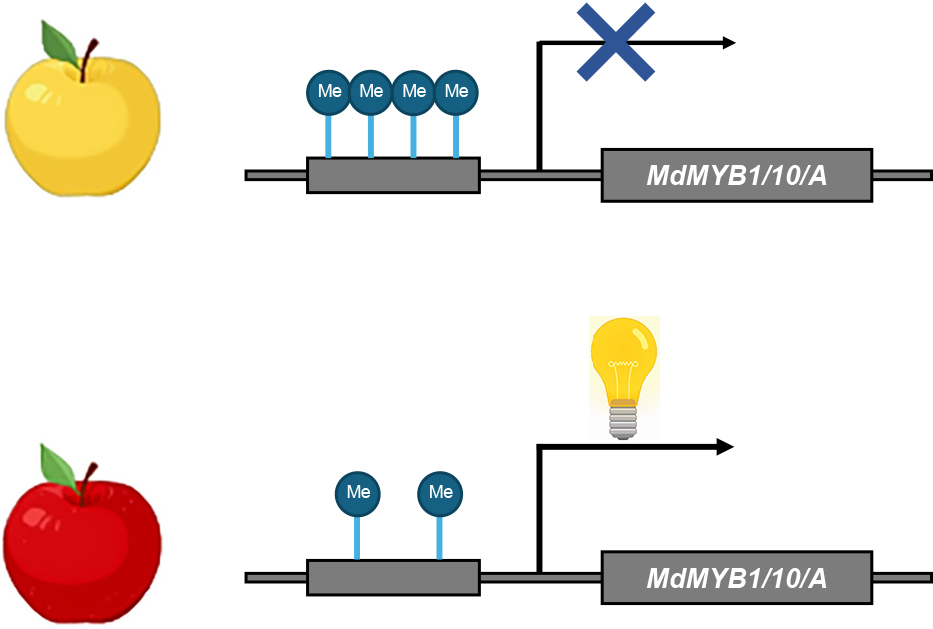
In fruit plants, somatic mutations or bud-sport mutations can alter the accumulation of anthocyanins, resulting in a change in the fruit skin color. Some of these changes are attributed to epigenetic mutations. In apples, changes in gene expression of the anthocyanin biosynthesis regulatory gene, MdMYB1/MdMYB10/MdMYBA, due to alteration in the DNA methylation state in the promoter region, lead to changes in anthocyanin accumulation and the redness of fruit skin (Fig. 5). For instance, in an anthocyanin-deficient yellow-skin apple mutant compared to its red-skin parent, the DNA methylation level in the promoter region of MdMYB1/MdMYB10/MdMYBA negatively correlated with the expression level and anthocyanin accumulation (Fig. 5) (El-Sharkawy et al., 2015). A bud-sport mutant exhibiting green to pale red fruit demonstrated a lower expression level of MdMYB1/MdMYB10/MdMYBA and a higher level of DNA methylation in the MdMYB1/MdMYB10/MdMYBA promoter region compared to the original plant with red fruit (Fig. 5) (Liu et al., 2022a). Similarly, a negative correlation between CHH methylation levels in the promoter region and MdMYB1/MdMYB10/MdMYBA expression levels or anthocyanin accumulation was observed among three different red-colored apple varieties possessing identical MdMYB1/MdMYB10/MdMYBA sequences (Fig. 5) (Jiang et al., 2020). Overexpression of MdAGO4 and MdDRM2 in apple callus resulted in increased CHH methylation of the MdMYB1/MdMYB10/MdMYBA promoter and decreased MdMYB1/MdMYB10/MdMYBA expression and anthocyanin accumulation (Jiang et al., 2020). Similarly, an association between DNA methylation of the promoter region of PcMYB1/PcMYB10/PcMYBA and PcMYB1/PcMYB10/PcMYBA gene expression and anthocyanin levels have been found in pears (Wang et al., 2013).
Environmental factors such as light or temperature can alter DNA methylation levels at the whole genome level, impacting the association between hypomethylation in the promoter region of genes encoding anthocyanin biosynthetic enzymes and upregulation in their expression levels, thereby affecting anthocyanin accumulation in the fruit of peach or skin of red pear (Liu et al., 2023; Zhu et al., 2020b).
In vegetables, certain Brassica vegetables have red or purple varieties, a result of selection for anthocyanin accumulation during the breeding process (Segawa et al., 2021). White-fleshed radish mutants derived from red-fleshed radishes exhibit reduced expression levels of RsMYB1. A CACTA transposon is present in the promoter region of RsMYB1, and there are no sequence differences in the RsMYB1 promoter, including the CACTA transposon, between white- and red-fleshed radishes. However, the DNA methylation level in the promoter region adjacent to the CACTA transposon was higher in white-fleshed radish than in red-fleshed radish. Additionally, anthocyanin accumulation and RsMYB1 expression were observed following treatment with 5-azaC. These results suggest that alternations in DNA methylation within the promoter region suppress RsMYB1 expression, resulting in white-fleshed radish mutants (Wang et al., 2020).
In ornamental plants such as Malus halliana, flower color transitions from red to white during development, are accompanied by decreased expression levels of structural genes including MhPAL, MhCHS, MhCHI, MhDFR, and MhANS. There is a positive correlation between the MhMYB10 expression level and red color intensity, as well as a negative correlation between MhMYB10 expression levels and DNA methylation levels within its promoter region during the transition period from red to white flowers (Han et al., 2020). Using foot bud cuttings from a heterochromatic chrysanthemum accession (Chrysanthemum morifolium), yellow and pink flower lines were generated. The pink flower line exhibited higher anthocyanin content in petals compared to the yellow flower line, along with elevated expression levels of CmMYB6, resulting in increased expression of downstream genes including CmF3H, CmDFR, CmANS, and Cm3GT. Although no sequence differences were detected in the CmMYB6 gene between the yellow and pink flower lines, CHH methylation levels were higher in the yellow line than in the pink flower line. Furthermore, demethylation of the promoter region in CmMYB6 using epigenome editing with the CRISPR-dCas-TET1cd demethylation system resulted in increased CmMYB6 expression and anthocyanin biosynthesis, suggesting that DNA methylation within the promoter region of CmMYB6 represses its expression, resulting in reduced anthocyanin levels and a yellow petal color (Tang et al., 2022).
Role of epigenetics under stress
Because plants are sessile organisms, they cannot avoid biotic and abiotic stresses, unlike animals. Therefore, plants have evolved complex mechanisms to cope with various biotic and abiotic stresses. Recent studies have revealed that epigenetic transcriptional regulation could be involved in plant adaptation under biotic and abiotic stress conditions. Increasingly, loss-of-function mutants of genes involved in epigenetic modification show altered responses to stress, or epigenetic modifications are altered under stress, indicating that epigenetic regulation plays an important role in stress responses (Chinnusamy and Zhu, 2009; Dowen et al., 2012).
In recent years, high- and low-temperature stress due to climate change has severely affected crop growth and yields worldwide. Plants have developed mechanisms to sense the surrounding temperature and regulate their metabolism and cellular functions to prevent damage caused by high and low temperatures (Fang et al., 2014; Miura and Furumoto, 2013). Variation in DNA methylation in the natural plant population of a clonal wild strawberry (Fragaria vesca) was examined by WGBS using samples from 21 natural populations across three climatically distinct geographic regions. Environmental factors (climate) have a greater impact on DNA methylation than genetic variation, and non-CG methylation was sensitive to climatic conditions. Variation in DNA methylation associated with climate was heritable across clonal generations, suggesting that heritable epigenetic changes induced by climate may be involved in adaptation to environmental conditions independent of genetic adaptation (Sammarco et al., 2024). In the wild strawberry F. vesca, DNA methylation levels decreased at most differentially methylated regions (DMRs) in genes, promoters, and TEs when subjected to heat stress (López et al., 2022). DMRs were also enriched in genes involved in the regulation and activity of transcription factors and the generation of metabolites and energy in response to heat stress (López et al., 2022). The DNA methylation pattern under 4 h and 12 h heat stress was examined in non-heading Chinese cabbage (B. rapa var. chinensis) by WGBS, and DMRs between the presence and absence of heat stress were identified. CG-related DMRs were identified, and there were a few CHH-related DMRs. DMRs were found in genic and intergenic regions, but there was less altered DNA methylation in TEs. The different gene ontology (GO) categories were overrepresented in genes associated with DMRs between 4 h and 12 h heat stress; switching of different sets of heat stress-induced genes was observed. Differences in DNA methylation levels in genic regions did not always cause changes in gene expression (Liu et al., 2018b). In broccoli, higher temperatures during floral development resulted in floral development cessation, producing cauliflower-like curd. The DNA methylation states in apexes at four different developmental stages were compared between different temperatures (28°C, 22°C, and 16°C) by WGBS, and a temperature-dependent increase in DNA methylation levels was observed, suggesting that it may be possible to grow broccoli in warmer regions by inhibiting increased DNA methylation due to warm temperatures through chemical treatment or using mutants of genes involved in DNA methylation (Yao et al., 2022). In rose, increased DNA methylation levels in response to low-temperature stress reduced expression levels of several key volatile biosynthesis-related enzymes, resulting in reduced emission of aromatic volatiles (Xie et al., 2023). Salt and drought stresses are known to inhibit plant growth and reduce overall productivity. Salt stress induces genome-wide changes in DNA methylation states, and different effects on DNA methylation were induced in diverse plant species and specific genes (Mastan et al., 2012). In olive, DNA methylation levels increased when plants were exposed to salt stress. These changes were more pronounced in salt-tolerant varieties (Mousavi et al., 2019). In tomato, the S-adenosylmethionine synthase (SlSAMS1), a core gene in the circadian rhythm pathway, was found to enhance salt tolerance by increasing DNA methylation levels of the gene body region of SlSAMS1 (Chen et al., 2023).
Changes in histone modification following stress treatment have also been investigated. In potato, distributions of H3K4me3 and H3K27me3 marks were compared between tubers stored at room temperature and low temperature. In tubers stored at cold temperature, both H3K4me3 and H3K27me3 marks accumulated throughout the gene body regions of active genes. However, this type of enrichment was not observed in tubers stored at room temperature. Sequential-ChIP showed that 30% of active genes with both H3K4me3 and H3K27me3 modifications had bivalent histone modifications. No association between the level of the bivalent histone modification and gene expression level was found when comparing tubers stored at room temperature and low temperature. However, bivalent-marked genes categorized into stress response tended to be upregulated in tubers stored at low temperatures, and bivalent-marked genes categorized into developmental and reproductive processes tended to be downregulated, suggesting that the chromatin structure produced by bivalent histone modification could be suitable for transcriptional response to cold stress in potato tubers (Zeng et al., 2019). In rose, HDAC expression was downregulated and HAT expression was upregulated following high-temperature stress, suggesting that enhancing histone acetylation levels may help roses cope with high-temperature stress (Wu et al., 2022).
The relationship between DNA methylation and disease resistance has also been studied. Loss-of-function mutants of genes involved in DNA methylation showed increased or decreased disease resistance (Tirnaz and Batley, 2019), suggesting that DNA methylation is also involved in disease resistance. Indeed, in A. thaliana, differences in the naturally occurring DNA methylation levels of two adjacent nucleotide-binding site and leucine-rich repeat (NLR) genes result in differences in clubroot resistance, and DNA methylation of these two genes that cause transcriptional silencing is maintained by DDM1, MET1, VIM, and CMT2/3 in the epiallele of a clubroot susceptible accession (Gravot et al., 2024). Furthermore, non-CG methylation tends to be higher in NLR genes with a fast evolutionary rate and high copy number, suggesting that DNA methylation is involved in the expansion and diversification of NLR genes (Cao et al., 2023). DNA methylation levels are altered by pathogen infection. In melon, DNA methylation levels were increased by Podosphaera xanthii infection, and about 30% of DEGs showed hyper- or hypomethylation (Wang et al., 2021). Only a few hundred DMRs were detected following Albugo candida infection in komatsuna (B. rapa var. perviridis) compared to non-infected plants. No overlapping DMRs were found between resistant and susceptible varieties, indicating DMRs in A. candida infection are variety-specific. Only 13 DMR-associated genes were differentially expressed, suggesting that the effect of DNA methylation changes caused by A. candida infection on expression is small (Tirnaz et al., 2022). Changes in DNA methylation due to insect herbivory have also been observed. In potato, DNA methylation levels were increased one day post-simulated herbivory with Phthorimaea operculella and then decreased, returning to non-treated levels at 14 days post-herbivory stimulation. Knocking down StDML2 by RNAi (StDML2i) resulted in decreased herbivory tolerance to P. operculella and reduced jasmonate biosynthesis. In StDML2i, the promoter region of jasmonate biosynthetic genes was hypermethylated. Higher DNA methylation states one day post-simulated herbivory of P. operculella were maintained in StDML2i, repressing transcription, while the wild type decreased DNA methylation levels, suggesting that StDML2 is involved in plant-herbivore interaction through jasmonate biosynthesis (Zhang et al., 2024). SlSDG33 and SlSDG34, possible orthologs of AtSDG8, are H3K4 and H3K36 methylase, and double mutants of slsdg33/slsdg34 showed increased disease resistance to the fungal pathogen, Botrytis cinerea. The double mutants of slsdg33/slsdg34 showed enhanced drought stress tolerance, suggesting that SlSDG33 and SlSDG34 activate transcription of negative regulators of biotic and abiotic stress tolerance via active histone modification in tomato (Bvindi et al., 2022).
Integrating epigenetic approaches into crop breeding practices
There are many natural epigenetic variants, such as epialleles, that can influence agricultural traits. These epigenetic changes that relate to desired traits can be defined by developing markers specific to that epigenetic state, termed epi-markers (Fig. 6). PCR-based techniques such as methylation-sensitive PCR or bisulfite PCR detect whether cytosines are methylated by observing changes in amplification patterns after treating DNA with bisulfite or digesting with methylation-sensitive restriction enzymes. The presence or absence of amplification, or changes in fragment size, reflect the methylation state, allowing identification of epigenetic polymorphisms linked to traits. These methods can be combined with high-throughput sequencing (NGS), enabling high-throughput and base-resolution analysis of methylation patterns across multiple loci or individuals, which is especially useful for identifying and validating epi-markers in crop populations. In maize, identified DMRs across inbreds show association with DMRs, but not to genomic SNPs (Xu et al., 2019), implying the unique value of epi-markers that cannot be detected by traditional genomic markers (Turcotte et al., 2022).
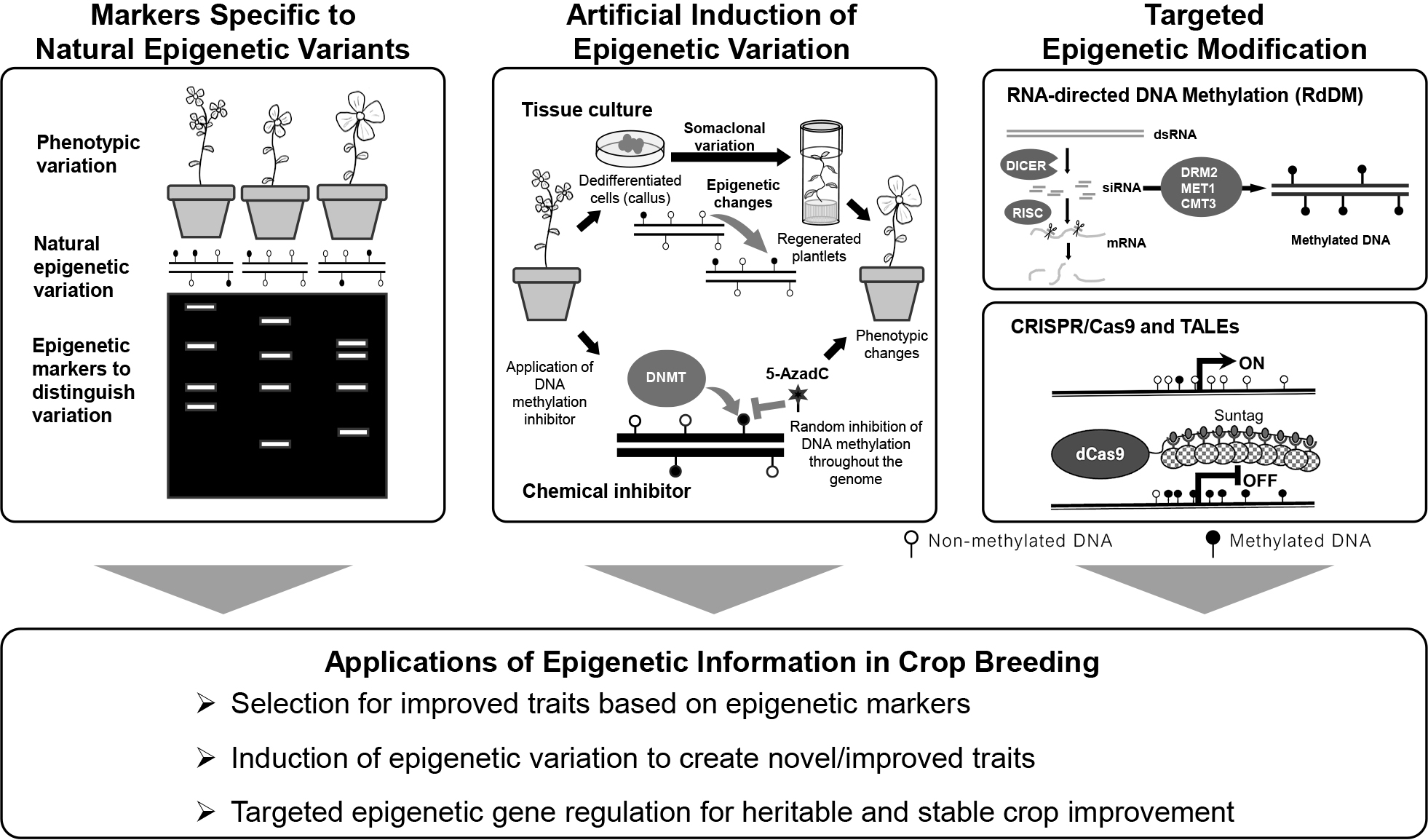
Artificially inducing epigenetic variation could potentially expand the phenotypic diversity that could be used for breeding purposes. Tissue culture and chemical induction induce DNA methylation changes (Fig. 6). Tissue culture induces an undifferentiated cell state (callus), which can be regenerated into plants by adjusting the hormone concentration in the plant growth medium. Plants regenerated from the same tissue harbor the same genome but can have different phenotypes accompanied by genetic and epigenetic changes, termed somaclonal variation. DNA methylation levels can increase depending on the hormone concentration (Azizi et al., 2020); however, heritable DNA hypomethylation has been observed in oil palm, maize, rice and A. thaliana (Ong-Abdullah et al., 2015; Stelpflug et al., 2014; Stroud et al., 2013; Tanurdzic et al., 2008). Thus, tissue culture is one approach to induce phenotypic variation that may be useful for breeding.
There are also chemicals that can induce epigenetic changes. A chemical inhibitor of DNA methylation has been used to modify the natural DNA methylation state that occurs within the genome (Fig. 6). Namely, 5′-aza-2′-deoxycytidine (5-AzadC) and Zebularine are cytosine analogs that replace cytosine during its incorporation into the genome. Inhibition of DNA methylation can be induced by germinating and growing the plants on a culture medium containing these inhibitors, or application of dissolved solutions (e.g., Chang et al., 2024; Finnegan et al., 2018). Depending on the application method, it is important to consider the cell division rate after incorporation of the inhibitors because the inhibition likely occurs during the DNA replication stage and the original DNA methylation state will diffuse over each cell division. The 5-AzadC forms a covalent bond with DNA methyltransferase but does not accept the transfer of the methyl group leading to inhibition of DNA methylation (Michalowsky and Jones, 1987). Zebularine inhibits DNA methylation by a similar mechanism (Champion et al., 2010; Cheng et al., 2003). The use of Zebularine on wheat (Triticum aestivum L.) showed a heritable increase in spikelet number, likely influenced by epigenetic changes (Finnegan et al., 2018). While DNA methylation inhibitors are useful for investigating the phenotypic effects induced by DNA methylation changes, the effects are often transient (Baubec et al., 2009) and can also induce genetic changes (Jackson-Grusby et al., 1997).
The above-mentioned methods rely on the identification of epigenetic changes that are linked to useful traits. Tissue culture and chemically induced epigenetic changes occur randomly within the genome and generally require genome-wide analysis of the epigenetic state and high-throughput screening for efficient identification of useful epi-markers. As an alternative method to use epigenetic information for breeding, a sequence-specific epigenome editing method has been developed. Gene silencing that depends on the recognition of nucleic acid sequence homology at the DNA or RNA level has been demonstrated in plants. It is currently thought that dsRNAs are involved in two types of gene silencing; 1) post-transcriptional gene silencing (PTGS), which involves targeted degradation of mRNA in the cytoplasm; 2) transcriptional gene silencing (TGS) at the genome level, induced by RdDM (described in Introduction and reviewed in Karimi and Innes, 2022).
RNAi refers to dsRNA-mediated gene silencing against mRNA with complementary sequences. This mechanism is present in plants, mammals, insects, and fungi (Elbashir et al., 2001; Fire et al., 1998; Mahmood et al., 2008). Through extensive genetic and biochemical analyses, it has become clear that gene silencing by RNAi involves a two-stage mechanism. In the first step, dsRNA is degraded by RNase III-like activity into siRNA of 21 to 25 nucleotides in length, by Dicer (Hutvágner et al., 2001; Lee et al., 2004). In the second step, siRNA binds to the RNA-induced silencing complex (RISC), an RNase complex, which acts on homologous mRNA leading to degradation. Several important components involved in RNAi, such as Dicer, RNA-dependent RNA polymerases, helicases, and dsRNA endonucleases, have been identified in a variety of organisms (Hammond et al., 2000; Martinez and Tuschl, 2004; Schwarz et al., 2003; Tomari et al., 2004). Targeted gene silencing through dsRNA-mediated RNAi has been widely applied. RNAi allows specific target genes to be selectively controlled, as opposed to the action of insecticides, which are frequently much broader in their efficacy (Fletcher et al., 2020). High-throughput, relatively low-cost, sequencing technologies, developed over the last 10–15 years, enable the determination of the genome sequences of target organisms. Application of RNAi as a successful control method requires knowledge of the sequences of candidate target loci which are required to design the dsRNAs.
There are several ways to express or deliver the dsRNA to induce RNAi in plants. Transfer of siRNAs from the dsRNA expressing scions towards the root stock via the plasmodesmata and sieve elements can be achieved by grafting. Grafting between an RNAi-induced tobacco (Nicotiana benthamiana) scion and potato (Solanum tuberosum) rootstock induced TGS via the RdDM pathway, and the TGS was maintained in the progeny lacking the siRNAs (Kasai et al., 2016). Using transgenic RNAi scions to induce DNA methylation of targeted loci in the non-transgenic root stocks may allow targeted gene regulation without transgene integration.
Alternatively, exogenous application of dsRNA has recently emerged as an appealing, non-GM technique to alter target gene expression (Koch et al., 2016; San Miguel and Scott, 2016). Initially, the practical application under field conditions of dsRNA application faced the challenge that dsRNA sprayed on a plant would not be sufficiently persistent due to UV degradation. The actin-dsRNA complex sprayed on leaves was not affected by UV degradation for at least 28 days under greenhouse conditions (where UV exposure level may be lower than under field conditions). Mitter et al. (2017a, b) invented clay nanosheets for effective protection of dsRNA on leaf surfaces under field conditions. Once loaded onto the nanosheets, dsRNA can be detected at 30 days after application. The clay is suitable for mass-production, meaning that spray-induced RNAi is a realistic tool to deploy against invertebrates and viruses in future pest and pathogen control strategies (Mitter et al., 2017b).
RdDM was first demonstrated in experiments with RNA viroids in tobacco, where RNA was able to cause cytosine methylation of identical genomic DNA sequences (Wassenegger et al., 1994). Wassenegger et al. (1994) generated tobacco plants with DNA sequences identical to the viroid in the genome of the integrated transgene. They found that these targeted transgene sequences were efficiently cytosine-methylated in plants with actively replicating viroids, but not in replication-deficient control plants. Plants utilize dsRNA produced by viruses, transgenes, and transposons to trigger various defense mechanisms, such as RNA degradation by RNAi and heterochromatin formation by cytosine methylation of the identical DNA sequence. How the RNA matches the DNA sequence and acquires cytosine methyltransferases remains unclear. However, the identification of cytosine methyltransferase as a mediator of non-CG methylation associated with RNA signaling provides a biochemical tool to isolate the relevant protein component. Thus, plant RdDM represents part of an RNA-based mechanism for targeted gene silencing in eukaryotes (Fig. 6).
Unlike induction of RdDM by dsRNA, DNA methylation can be directly induced by localizing a DNA methyltransferase enzyme to the target DNA locus. Transcription activator-like effectors (TALEs) and CRISPR/Cas9 have been used for targeted epigenome editing. TALEs are naturally occurring transcriptional activators secreted by the plant pathogen Xanthomonas spp., and by fusing a DNA methylation enzyme such as DNA methyltransferase 3 alpha (DNMT3A) or ten-eleven translocation 1 (TET1), DNA methylation modification in mammalian cells has been demonstrated (Lo et al., 2017). Cas9 is a DNA endonuclease that associates with a single-guide RNA (sgRNA) consisting of a 20-nucleotide recognition sequence. Cas9/sgRNA complex is directed to bind to the genomic DNA sequence that is complementary to the sgRNA recognition sequence, which cleaves and induces mutations at or near the binding site (Fig. 6). A dead Cas9 (dCas9) is a mutant of Cas9 that will still bind to the target DNA but will not cleave the site. Similar to TALEs, an epigenomic modifying enzyme fused to dCas9 can direct epigenome modification of the target region. Indeed, CHG methylation by DRM2 (Papikian et al., 2019) and CG methylation by MQ1 (Ghoshal et al., 2021) have been demonstrated in plants. Recently, dCas9 epigenome modification using multiple modifiers (e.g. DNA methyltransferase and H3K27me3) has been developed for mammalian systems (Swain et al., 2024), which may be useful for multi-layered epigenome modification that may lead to more stable gene regulation. The dCas9 transgenes will need to be transformed into a plant species of interest for targeted epigenome editing and subsequently crossed to a non-transgenic plant to segregate out the dCas9 transgenes to create a non-transgenic epigenome modified plant. However, understanding the stability and heritability of the edited epigenome in different plant species will require further research.
Perspectives
Research focused on the role of epigenetics in horticultural plants has been increasing (Fig. 7). Breakthroughs in whole genome sequencing now make it possible to conduct in-depth comprehensive epigenetic analyses across diverse species. However, WGBS remains challenging for species with large genome sizes and high ploidy, and approaches such as reduced representation bisulfite sequencing (RRBS) have been developed. Advances in epigenome analysis have led to greater interest in understanding how epigenetic variations influence plant traits. Some traits selected during the breeding process may result from epimutations. Exploring how epigenetic modifications evolve in long-lived plants, such as fruit trees, could produce insights into their resilience and adaptability over decades. As climate change continues to impact horticultural crop production, emerging evidence suggests a link between environmental changes and epigenetic modifications. This raises the possibility that environmental stimuli may induce heritable epimutations, potentially aiding in crop adaptation. While much of the current research shows correlation between epigenetic modifications and changes in gene expression, cutting-edge techniques in epigenome editing are beginning to demonstrate these effects more directly. Driven by rapid technological advancements and increasing evidence on the crucial role of epigenetics, future research in horticultural plant epigenetics promises to unlock new strategies for enhancing crop resilience, quality, and productivity.
Literature Cited
- Abhinandan, K., S. Sankaranarayanan, S. Macgregor, D. R. Goring and M. A. Samuel. 2022. Cell-cell signaling during the Brassicaceae self-incompatibility response. Trends. Plant Sci. 27: 472–487.
- Akagi, T., I. M. Henry, R. Tao and L. Comai. 2014. A Y-chromosome encoded small RNA acts as a sex determinant in persimmons. Science 346: 646–650.
- Akagi, T., I. M. Henry, T. Kawai, L. Comai and R. Tao. 2016. Epigenetic regulation of the sex determination gene MeGI in polyploid persimmon. Plant Cell 28: 2905–2915.
- Akter, A., E. Itabashi, T. Kakizaki, K. Okazaki, E. S. Dennis and R. Fujimoto. 2021. Genome triplication leads to transcriptional divergence of FLOWERING LOCUS C genes during vernalization in the genus Brassica. Front. Plant Sci. 11: 619417. DOI: 10.3389/fpls.2020.619417.
- Akter, A., J. Miyazaki, D. J. Shea, N. Nishida, S. Takada, N. Miyaji, H. Mehraj, M. Shimizu, M. A. U. Doullah, T. Takasaki-Yasuda, K. Okazaki and R. Fujimoto. 2020. Gene expression analysis in response to vernalization in Chinese cabbage (Brassica rapa L.). Hort. J. 89: 268–277.
- Akter, A., S. Takahashi, W. Deng, D. J. Shea, E. Itabashi, M. Shimizu, N. Miyaji, K. Osabe, N. Nishida, Y. Suzuki, C. A. Helliwell, M. Seki, W. J. Peacock, E. S. Dennis and R. Fujimoto. 2019. The histone modification H3 lysine 27 tri-methylation has conserved gene regulatory roles in the triplicated genome of Brassica rapa L. DNA Res. 26: 433–443.
- Akter, M. A., H. Mehraj, N. Miyaji, S. Takahashi, T. Takasaki-Yasuda, M. Seki, E. S. Dennis, R. Fujimoto and K. Osabe. 2022. Transcriptional association between mRNAs and their paired natural antisense transcripts following Fusarium oxysporum inoculation in Brassica rapa L. Horticulturae 8: 17. DOI: 10.3390/horticulturae8010017.
- Azeez, A., Y. C. Zhao, R. K. Singh, Y. S. Yordanov, M. Dash, P. Miskolczi, K. Stojkovič, S. H. Strauss, R. P. Bhalerao and V. B. Busov. 2021. EARLY BUD-BREAK 1 and EARLY BUD-BREAK 3 control resumption of poplar growth after winter dormancy. Nat. Commun. 12: 1123. DOI: 10.1038/s41467-021-21449-0.
- Azizi, P., M. M. Hanafi, M. Sahebi, J. A. Harikrishna, S. Taheri, A. Yassoralipour and A. Nasehi. 2020. Epigenetic changes and their relationship to somaclonal variation: a need to monitor the micropropagation of plantation crops. Funct. Plant Biol. 47: 508–523.
- Bateman, A. J. 1955. Self-incompatibility systems in angiosperms. III. Cruciferae. Heredity 9: 53–68.
- Baubec, T., A. Pecinka, W. Rozhon and O. Mittelsten Scheid. 2009. Effective, homogeneous and transient interference with cytosine methylation in plant genomic DNA by zebularine. Plant J. 57: 542–554.
- Berry, S. and C. Dean. 2015. Environmental perception and epigenetic memory: mechanistic insight through FLC. Plant J. 83: 133–148.
- Boureau, L., A. How-Kit, E. Teyssier, S. Drevensek, M. Rainieri, J. Joubès, L. Stammitti, A. Pribat, C. Bowler, Y. Hong and P. Gallusci. 2016. A CURLY LEAF homologue controls both vegetative and reproductive development of tomato plants. Plant Mol. Biol. 90: 485–501.
- Bräutigam, K., R. Soolanayakanahally, M. Champigny, S. Mansfield, C. Douglas, M. M. Campbell and Q. Cronk. 2017. Sexual epigenetics: gender-specific methylation of a gene in the sex determining region of Populus balsamifera. Sci. Rep. 7: 45388. DOI: 10.1038/srep45388.
- Bvindi, C., S. Lee, L. Tang, M. V. Mickelbart, Y. Li and T. Mengiste. 2022. Improved pathogen and stress tolerance in tomato mutants of SET domain histone 3 lysine methyltransferases. New Phytol. 235: 1957–1976.
- Candela-Ferre, J., B. Diego-Martin, J. Pérez-Alemany and J. Gallego-Bartolomé. 2024. Mind the gap: Epigenetic regulation of chromatin accessibility in plants. Plant Physiol. 194: 1998–2016.
- Canton, M., S. Farinati, C. Forestan, J. Joseph, C. Bonghi and S. Varotto. 2022. An efficient chromatin immunoprecipitation (ChIP) protocol for studying histone modifications in peach reproductive tissues. Plant Methods 18: 43.
- Cao, Q., S. Wang, H. Ma, S. Luo, H. He and Y. Zhang. 2023. Hyper non-CG methylation of expanded plant disease resistance NLR genes. Plant Cell Rep. 42: 1251–1254.
- Cermak, T., Z. Kubat, R. Hobza, A. Koblizkova, A. Widmer, J. Macas, B. Vyskot and E. Kejnovsky. 2008. Survey of repetitive sequences in Silene latifolia with respect to their distribution on sex chromosomes. Chromosome Res. 16: 961–976.
- Champion, C., D. Guianvarc’h, C. Senamaud-Beaufort, R. Z. Jurkowska, A. Jeltsch, L. Ponger, P. B. Arimondo and A. L. Guieysse-Peugeot. 2010. Mechanistic insights on the inhibition of C5 DNA methyltransferases by zebularine. PLoS One 5: e12388. DOI: 10.1371/journal.pone.0012388.
- Chang, X., L. Liu, Z. Liu, L. Qiao, R. Shi and L. Lu. 2024. Chemical induction of DNA demethylation by 5-Azacytidine enhances tomato fruit defense against gray mold through dicer-like protein DCL2c. Hortic Res. 11: uhae164. DOI: 10.1093/hr/uhae164.
- Chen, J., J. Cheng, X. Chen, M. Inoue, Y. Liu and C. X. Song. 2022a. Whole-genome long-read TAPS deciphers DNA methylation patterns at base resolution using PacBio SMRT sequencing technology. Nucleic Acids Res. 50: e104. DOI: 10.1093/nar/gkac612.
- Chen, W., Y. Tamada, H. Yamane, M. Matsushita, Y. Osako, M. Gao-Takai, Z. Luo and R. Tao. 2022b. H3K4me3 plays a key role in establishing permissive chromatin states during bud dormancy and bud break in apple. Plant J. 111: 1015–1031.
- Chen, X., G. Chen, S. Guo, Y. Wang and J. Sun. 2023. SlSAMS1 enhances salt tolerance through regulation DNA methylation of SlGI in tomato. Plant Sci. 335: 111808. DOI: 10.1016/j.plantsci.2023.111808.
- Cheng, J. C., C. B. Matsen, F. A. Gonzales, W. Ye, S. Greer, V. E. Marquez, P. A. Jones and E. U. Selker. 2003. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J. Natl. Cancer Inst. 95: 399–409.
- Cheng, J., Q. Niu, B. Zhang, K. Chen, R. Yang, J. K. Zhu, Y. Zhang and Z. Lang. 2018. Downregulation of RdDM during strawberry fruit ripening. Genome Biol. 19: 212.
- Chinnusamy, V. and J. K. Zhu. 2009. Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 12: 133–139.
- Corem, S., A. Doron-Faigenboim, O. Jouffroy, F. Maumus, T. Arazi and N. Bouché. 2018. Redistribution of CHH methylation and small interfering RNAs across the genome of tomato ddm1 mutants. Plant Cell 30: 1628–1644.
- de la Fuente, L., A. Conesa, A. Lloret, M. L. Badenes and G. Ríos. 2015. Genome-wide changes in histone H3 lysine 27 trimethylation associated with bud dormancy release in peach. Tree Genet. Genomes 11: 45. DOI: 10.1007/s11295-015-0869-7.
- Ding, X., X. Liu, G. Jiang, Z. Li, Y. Song, D. Zhang, Y. Jiang and X. Duan. 2022. SlJMJ7 orchestrates tomato fruit ripening via crosstalk between H3K4me3 and DML2-mediated DNA demethylation. New Phytol. 233: 1202–1219.
- Dowen, R. H., M. Pelizzola, R. J. Schmitz, R. Lister, J. M. Dowen, J. R. Nery, J. E. Dixon and J. R. Ecker. 2012. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 109: E2183–E2191.
- Durand, E., R. Mèheust, M. Soucaze, P. M. Goubet, S. Gallina, C. Poux, I. Fobis-Loisy, E. Guillon, T Gaude, A. Sarazin, M. Figeac, E. Prat, W. Marande, H. Bergès, X. Vekemans, S. Billiard and V. Castric. 2014. Dominance hierarch arising from the evolution of a complex small RNA regulatory network. Science 346: 1200–1205.
- Elbashir, S. M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498.
- El-Sharkawy, I., D. Liang and K. Xu. 2015. Transcriptome analysis of an apple (Malus × domestica) yellow fruit somatic mutation identifies a gene network module highly associated with anthocyanin and epigenetic regulation. J. Exp. Bot. 66: 7359–7376.
- Fang, H., X. Liu, G. Thorn, J. Duan and L. Tian. 2014. Expression analysis of histone acetyltransferases in rice under drought stress. Biochem. Biophys. Res. Commun. 443: 400–405.
- Finnegan, E. J., B. Ford, X. Wallace, F. Pettolino, P. T. Griffin, R. J. Schmitz, P. Zhang, J. M. Barrero, M. J. Hayden, S. A. Boden, C. A. Cavanagh, S. M. Swain and B. Trevaskis. 2018. Zebularine treatment is associated with deletion of FT-B1 leading to an increase in spikelet number in bread wheat. Plant Cell Environ. 41: 1346–1360.
- Fire, A., S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello. 1998. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806–811.
- Fletcher, S. J., P. T. Reeves, B. T. Hoang and N. Mitter. 2020. A perspective on RNAi-based biopesticides. Front. Plant Sci. 11: 51. DOI: 10.3389/fpls.2020.00051.
- Fu, W., S. Huang, Y. Gao, M. Zhang, G. Qu, N. Wang, Z. Liu and H. Feng. 2020. Role of BrSDG8 on bolting in Chinese cabbage (Brassica rapa). Theor. Appl. Genet. 133: 2937–2948.
- Fuchs, J., D. Demidov, A. Houben and I. Schubert. 2006. Chromosomal histone modification patterns—from conservation to diversity. Trends Plant Sci. 11: 199–208.
- Fujii, S. and S. Takayama. 2018. Multilayered dominance hierarchy in plant self-incompatibility. Plant Reprod. 31: 15–19.
- Fujimoto, R., J. M. Taylor, S. Shirasawa, W. J. Peacock and E. S. Dennis. 2012b. Heterosis of Arabidopsis hybrids between C24 and Col is associated with increased photosynthesis capacity. Proc. Natl. Acad. Sci. USA 109: 7109–7114.
- Fujimoto, R., K. Uezono, S. Ishikura, K. Osabe, W. J. Peacock and E. S. Dennis. 2018. Recent research on the mechanism of heterosis is important for crop and vegetable breeding systems. Breed. Sci. 68: 145–158.
- Fujimoto, R., T. Sasaki, H. Inoue and T. Nishio. 2008b. Hypomethylation and transcriptional reactivation of retrotransposon-like sequences in ddm1 transgenic plants of Brassica rapa. Plant Mol. Biol. 66: 463–473.
- Fujimoto. R., T. Sasaki, H. Kudoh, J. M. Taylor, T. Kakutani and E. S. Dennis. 2011. Epigenetic variation in the FWA gene within the genus Arabidopsis. Plant J. 66: 831–843.
- Fujimoto, R., T. Sasaki, R. Ishikawa, K. Osabe, T. Kawanabe and E. S. Dennis. 2012a. Molecular mechanisms of epigenetic variation in plants. Int. J. Mol. Sci. 13: 9900–9922.
- Fujimoto, R., T. Sugimura, E. Fukai and T. Nishio. 2006. Suppression of gene expression of a recessive SP11/SCR allele by an untranscribed SP11/SCR allele in Brassica self-incompatibility. Plant Mol. Biol. 61: 753–765.
- Fujimoto, R., Y. Kinoshita, A. Kawabe, T. Kinoshita, K. Takashima, M. Nordborg, M. E. Nasrallah, K. K. Shimizu, H. Kudoh and T. Kakutani. 2008a. Evolution and control of imprinted FWA genes in the genus Arabidopsis. PLoS Genet. 4: e1000048. DOI: 10.1371/journal.pgen.1000048.
- Furey, T. S. 2012. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat. Rev. Genet. 13: 840–852.
- Gao, Y., Q. Yang, X. Yan, X. Wu, F. Yang, J. Li, J. Wei, J. Ni, M. Ahmad, S. Bai and Y. Teng. 2021. High-quality genome assembly of ‘Cuiguan’ pear (Pyrus pyrifolia) as a reference genome for identifying regulatory genes and epigenetic modifications responsible for bud dormancy. Hortic. Res. 8: 197. DOI: 10.1038/s41438-021-00632-w.
- Ghoshal, B., C. L. Picard, B. Vong, S. Feng and S. E. Jacobsen. 2021. CRISPR-based targeting of DNA methylation in Arabidopsis thaliana by a bacterial CG-specific DNA methyltransferase. Proc. Natl. Acad. Sci. USA 118: e2125016118. DOI: 10.1073/pnas.2125016118.
- Goring, D. R., M. Bosch and V. E. Franklin-Tong. 2023. Contrasting self-recognition rejection systems for self-incompatibility in Brassica and Papaver. Curr. Biol. 33: R530–R542.
- Gouil, Q. and D. C. Baulcombe. 2016. DNA methylation signatures of the plant chromomethyltransferases. PLoS Genet. 12: e1006526. DOI: 0.1371/journal.pgen.1006526.
- Gravot, A., B. Liégard, L. Quadrana, F. Veillet, Y. Aigu, T. Bargain, J. Bénéjam, C. Lariagon, J. Lemoine, V. Colot, M. J. Manzanares-Dauleux and M. Jubault. 2024. Two adjacent NLR genes conferring quantitative resistance to clubroot disease in Arabidopsis are regulated by a stably inherited epiallelic variation. Plant Commun. 5: 100824. DOI: 10.1016/j.xplc.2024.100824.
- Grover, J. W., T. Kendall, A. Baten, D. Burgess, M. Freeling, G. J. King and R. A. Mosher. 2018. Maternal components of RNA-directed DNA methylation are required for seed development in Brassica rapa. Plant J. 94: 575–582.
- Gu, T., M. Mazzurco, W. Sulaman, D. D. Matias and D. R. Goring. 1998. Binding of an arm repeat protein to the kinase domain of the S-locus receptor kinase. Proc. Natl. Acad. Sci. USA 95: 382–387.
- Hammond, S. M., E. Bernstein, D. Beach and G. J. Hannon. 2000. An RNA-directed nuclease mediates posttranscriptional gene silencing in Drosophila cells. Nature 404: 293–296.
- Han, M. L., J. Yin, Y. H. Zhao, X. W. Sun, J. X. Meng, J. Zhou, T. Shen, H. H. Li and F. Zhang. 2020. How the color fades from Malus halliana flowers: Transcriptome sequencing and DNA methylation analysis. Front. Plant Sci. 11: 576054. DOI: 10.3389/fpls.2020.576054.
- Hatakeyama. K., M. Watanabe, T. Takasaki, K. Ojima and K. Hinata. 1998. Dominance relationships between S-alleles in self-incompatible Brassica campestris L. Heredity 80: 241–247.
- He, G., A. A. Elling and X. W. Deng. 2011. The epigenome and plant development. Annu. Rev. Plant Biol. 62: 411–435.
- Hermawaty, D., J. Cahn, R. Lister and M. J. Considine. 2023. Systematic evaluation of chromatin immunoprecipitation sequencing to study histone occupancy in dormancy transitions of grapevine buds. Tree Physiol. 43: 675–689.
- Hu, G., B. Huang, K. Wang, P. Frasse, E. Maza, A. Djari, M. Benhamed, P. Gallusci, Z. Li, M. Zouine and M. Bouzayen. 2021. Histone posttranslational modifications rather than DNA methylation underlie gene reprogramming in pollination-dependent and pollination-independent fruit set in tomato. New Phytol. 229: 902–919.
- Huang, H., R. Liu, Q. Niu, K. Tang, B. Zhang, H. Zhang, K. Chen, J. K. Zhu and Z. Lang. 2019. Global increase in DNA methylation during orange fruit development and ripening. Proc. Natl. Acad. Sci. USA 116: 1430–1436.
- Huang, J., L. Yang, L. Yang, X. Wu, X. Cui, L. Zhang, J. Hui, Y. Zhao, H. Yang, S. Liu, Q. Xu, M. Pang, X. Guo, Y. Cao, Y. Chen, X. Ren, J. Lv, J. Yu, J. Ding, G. Xu, N. Wang, X. Wei, Q. Lin, Y. Yuan, X. Zhang, C. Ma, C. Dai, P. Wang, Y. Wang, F. Cheng, W. Zeng, R. Palanivelu, H. M. Wu, X. Zhang, A. Y. Cheung and Q. Duan. 2023. Stigma receptors control intraspecies and interspecies barriers in Brassicaceae. Nature 614: 303–308.
- Huang, S., L. Hou, W. Fu, Z. Liu, C. Li, X. Li and H. Feng. 2020b. An insertion mutation in Bra032169 encoding a histone methyltransferase is responsible for early bolting in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Front. Plant Sci. 11: 547. DOI: 10.3389/fpls.2020.00547.
- Huang, X., Q. Pan, Y. Lin, T. Gu and Y. Li. 2020a. A native chromatin immunoprecipitation (ChIP) protocol for studying histone modifications in strawberry fruits. Plant Methods 16: 10. DOI: 10.1186/s13007-020-0556-z.
- Hutvágner, G., J. McLachlan, A. E. Pasquinelli, E. Bálint, T. Tuschl and P. D. Zamore. 2001. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293: 834–838.
- Indriolo, E., P. Tharmapalan, S. I. Wright and D. R. Goring. 2012. The ARC1 E3 ligase gene is frequently deleted in self-compatible Brassicaceae species and has a conserved role in Arabidopsis lyrata self-pollen rejection. Plant Cell 24: 4607–4620.
- Jackson-Grusby, L., P. W. Laird, S. N. Magge, B. J. Moeller and R. Jaenisch. 1997. Mutagenicity of 5-aza-2′-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc. Natl. Acad. Sci. USA 94: 4681–4685.
- Jiang, S., N. Wang, M. Chen, R. Zhang, Q. Sun, H. Xu, Z. Zhang, Y. Wang, X. Sui, S. Wang, H. Fang, W. Zuo, M. Su, J. Zhang, Z. Fei and X. Chen. 2020. Methylation of MdMYB1 locus mediated by RdDM pathway regulates anthocyanin biosynthesis in apple. Plant Biotechnol. J. 18: 1736–1748.
- Julian, R., R. M. Patrick and Y. Li. 2023. Organ-specific characteristics govern the relationship between histone code dynamics and transcriptional reprogramming during nitrogen response in tomato. Commun. Biol. 6: 1225. DOI: 10.1038/s42003-023-05601-8.
- Kakizaki, T., Y. Takada, A. Ito, G. Suzuki, H. Shiba, S. Takayama, A. Isogai and M. Watanabe. 2003. Linear dominance relationship among four class-II S haplotype in pollen is determined by the expression of SP11 in Brassica self-incompatibility. Plant Cell Physiol. 44: 70–75.
- Kakizaki, T., Y. Takada, T. Fujioka, G. Suzuki, Y. Satta, H. Shiba, A. Isogai, S. Takayama and M. Watanabe. 2006. Comparative analysis of the S-intergenic region in class-II S haplotypes of self-incompatible Brassica rapa (syn. campestris). Genes Genet. Syst. 81: 63–67.
- Kakutani, T., J. A. Jeddeloh, S. K. Flowers, K. Munakata and E. J. Richards. 1996. Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proc. Natl. Acad. Sci. USA 93: 12406–12411.
- Karimi, H. Z. and R. W. Innes. 2022. Molecular mechanisms underlying host-induced gene silencing. Plant Cell 34: 3183–3199.
- Kasai, A., S. Bai, H. Hojo and T. Harada. 2016. Epigenome editing of potato by grafting using transgenic tobacco as siRNA donor. PLoS One 11: e0161729. DOI: 10.1371/journal.pone.0161729.
- Kawakatsu, T. and J. R. Ecker. 2019. Diversity and dynamics of DNA methylation: epigenomic resources and tools for crop breeding. Breed. Sci. 69: 191–204.
- Kawanabe, T., S. Ishikura, N. Miyaji, T. Sasaki, L. M. Wu, E. Itabashi, S. Takada, M. Shimizu, T. Takasaki-Yasuda, K. Osabe, W. J. Peacock, E. S. Dennis and R. Fujimoto. 2016a. Role of DNA methylation in hybrid vigor in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 113: E6704–E6711.
- Kawanabe, T., K. Osabe, E. Itabashi, K. Okazaki, E. S. Dennis and R. Fujimoto. 2016b. Development of primer sets that can verify the enrichment of histone modifications, and their application to examining vernalization-mediated chromatin changes in Brassica rapa L. Genes Genet. Syst. 91: 1–10.
- Koch, A., D. Biedenkopf, A. Furch, L. Weber, O. Rossbach, E. Abdellatef, L. Linicus, J. Johannsmeier, L. Jelonek, A. Goesmann, V. Cardoza, J. McMillan, T. Mentzel and K. H. Kogel. 2016. An RNAi-based control of Fusarium graminearum infections through spraying of long dsRNAs involves a plant passage and is controlled by the fungal silencing machinery. PLoS Pathog. 12: e1005901. DOI: 10.1371/journal.ppat.1005901.
- Kubat, Z., J. Zluvova, I. Vogel, V. Kovacova, T. Cermak, R. Cegan, R. Hobza, B. Vyskot and E. Kejnovsky. 2014. Possible mechanisms responsible for absence of a retrotransposon family on a plant Y chromosome. New Phytol. 202: 662–678.
- Lai, Y. S., X. Zhang, W. Zhang, D. Shen, H. Wang, Y. Xia, Y. Qiu, J. Song, C. Wang and X. Li. 2017. The association of changes in DNA methylation with temperature-dependent sex determination in cucumber. J. Exp. Bot. 68: 2899–2912.
- Lang, Z., Y. Wang, K. Tang, D. Tang, T. Datsenka, J. Cheng, Y. Zhang, A. K. Handa and J. K. Zhu. 2017. Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. USA 114: 4511–4519.
- Latrasse, D., N. Y. Rodriguez-Granados, A. Veluchamy, K. G. Mariappan, C. Bevilacqua, N. Crapart, C. Camps, V. Sommard, C. Raynaud, C. Dogimont, A. Boualem, M. Benhamed and A. Bendahmane. 2017. The quest for epigenetic regulation underlying unisexual flower development in Cucumis melo. Epigenetics Chromatin 10: 22. DOI: 10.1186/s13072-017-0132-6.
- Lee, Y. S., K. Nakahara, J. W. Pham, K. Kim, Z. He, E. J. Sontheimer and R. W. Carthew. 2004. Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell 117: 69–81.
- Li, H., J. Yuan, M. Wu, Z. Han, L. Li, H. Jiang, Y. Jia, X. Han, M. Liu, D. Sun, C. Chen, W. Song and C. Wang. 2018. Transcriptome and DNA methylome reveal insights into yield heterosis in the curds of broccoli (Brassica oleracea L. var. italic). BMC Plant Biol. 18: 168. DOI: 10.1186/s12870-018-1384-4.
- Li, S. F., C. C. Lv, L. N. Lan, K. L. Jiang, Y. L. Zhang, N. Li, C. L. Deng and W. J. Gao. 2021. DNA methylation is involved in sexual differentiation and sex chromosome evolution in the dioecious plant garden asparagus. Hortic. Res. 8: 198. DOI: 10.1038/s41438-021-00633-9.
- Li, X., S. Zhang, J. Bai and Y. He. 2016. Tuning growth cycles of Brassica crops via natural antisense transcripts of BrFLC. Plant Biotechnol. J. 14: 905–914.
- Li, Y., Y. Shi, Y. Li, J. Lu, Y. Sun, Y. Zhang, W. Chen, X. Yang, D. Grierson, Z. Lang, G. Jiang and K. Chen. 2023. DNA methylation mediated by RdDM pathway and demethylation affects furanone accumulation through regulation of QUINONE OXIDOREDUCTASE in strawberry. Hortic. Res. 10: uhad131. DOI: 10.1093/hr/uhad131.
- Li, Z., G. Jiang, X. Liu, X. Ding, D. Zhang, X. Wang, Y. Zhou, H. Yan, T. Li, K. Wu, Y. Jiang and X. Duan. 2020. Histone demethylase SlJMJ6 promotes fruit ripening by removing H3K27 methylation of ripening-related genes in tomato. New Phytol. 227: 1138–1156.
- Liang, Q., H. Deng, Y. Li, Z. Liu, P. Shu, R. Fu, Y. Zhang, J. Pirrello, Y. Zhang, D. Grierson, M. Bouzayen, Y. Liu and M. Liu. 2020. Like Heterochromatin Protein 1b represses fruit ripening via regulating the H3K27me3 levels in ripening-related genes in tomato. New Phytol. 227: 485–497.
- Liu, D. D., L. J. Zhou, M. J. Fang, Q. L. Dong, X. H. An, C. X. You and Y. J. Hao. 2016. Polycomb-group protein SlMSI1 represses the expression of fruit-ripening genes to prolong shelf life in tomato. Sci. Rep. 6: 31806. DOI: 10.1038/srep31806.
- Liu, G., Y. Xia, T. Liu, S. Dai and X. Hou. 2018a. The DNA methylome and association of differentially methylated regions with differential gene expression during heat stress in Brassica rapa. Int. J. Mol. Sci. 19: 1414. DOI: 10.3390/ijms19051414.
- Liu, H. N., Q. Shu, K. Lin-Wang, R. V. Espley, A. C. Allan, M. S. Pei, X. L. Li, J. Su and J. Wu. 2023. DNA methylation reprogramming provides insights into light-induced anthocyanin biosynthesis in red pear. Plant Sci. 326: 111499. DOI: 10.1016/j.plantsci.2022.111499.
- Liu, M., J. Pirrello, C. Chervin, J. P. Roustan and M. Bouzayen. 2015a. Ethylene control of fruit ripening: Revisiting the complex network of transcriptional regulation. Plant Physiol. 169: 2380–2390.
- Liu, R., A. How-Kit, L. Stammitti, E. Teyssier, D. Rolin, A. Mortain-Bertrand, S. Halle, M. Liu, J. Kong, C. Wu, C. Degraeve-Guibault, N. H. Chapman, M. Maucourt, T. C. Hodgman, J. Tost, M. Bouzayen, Y. Hong, G. B. Seymour, J. J. Giovannoni and P. Gallusci. 2015b. A DEMETER-like DNA demethylase governs tomato fruit ripening. Proc. Natl. Acad. Sci. USA 112: 10804–10809.
- Liu, T., W. Duan, Z. Chen, J. Yuan, D. Xiao, X. Hou and Y. Li. 2020. Enhanced photosynthetic activity in pak choi hybrids is associated with increased grana thylakoids in chloroplasts. Plant J. 103: 2211–2224.
- Liu, W., S. H. Duttke, J. Hetzel, M. Groth, S. Feng, J. Gallego-Bartolome, Z. Zhong, H. Y. Kuo, Z. Wang, J. Zhai, J. Chory and S. E. Jacobsen. 2018b. RNA-directed DNA methylation involves co-transcriptional small-RNA guided slicing of polymerase V transcripts in Arabidopsis. Nat. Plants 4: 181–188.
- Liu, Y., X. H. Gao, L. Tong, M. Z. Liu, X. K. Zhou, M. M. Tahir, L. B. Xing, J. J. Ma, N. An, C. P. Zhao, J. L. Yao and D. Zhang. 2022a. Multi-omics analyses reveal MdMYB10 hypermethylation being responsible for a bud sport of apple fruit color. Hortic. Res. 9: uhac179. DOI: 10.1093/hr/uhac179.
- Liu, Z., X. Wu, H. Liu, M. Zhang and W. Liao. 2022b. DNA methylation in tomato fruit ripening. Physiol. Plant 174: e13627. DOI: 10.1111/ppl.13627.
- Lloyd, J. P. B. and R. Lister. 2022. Epigenome plasticity in plants. Nat. Rev. Genet. 23: 55–68.
- Lo, C. L., S. R. Choudhury, J. Irudayaraj and F. C. Zhou. 2017. Epigenetic editing of Ascl1 gene in neural stem cells by optogenetics. Sci. Rep. 7: 42047. DOI: 10.1038/srep42047.
- Long, J., J. Liu, A. Xia, N. M. Springer and Y. He. 2021. Maize decrease in DNA methylation 1 targets RNA-directed DNA methylation on active chromatin. Plant Cell 33: 2183–2196.
- López, M. E., D. Roquis, C. Becker, B. Denoyes and E. Bucher. 2022. DNA methylation dynamics during stress response in woodland strawberry (Fragaria vesca). Hortic. Res. 9: uhac174. DOI: 10.1093/hr/uhac174.
- Lü, P., S. Yu, N. Zhu, Y. R. Chen, B. Zhou, Y. Pan, D. Tzeng, J. P. Fabi, J. Argyris, J. Garcia-Mas, N. Ye, J. Zhang, D. Grierson, J. Xiang, Z. Fei, J. Giovannoni and S. Zhong. 2018. Genome encode analyses reveal the basis of convergent evolution of fleshy fruit ripening. Nat. Plants 4: 784–791.
- Mahmood, R., I. Ali, T. Husnain and S. Riazuddin. 2008. RNA interference: The story of gene silencing in plants and humans. Biotechnol. Adv. 26: 202–209.
- Manning, K., M. Tör, M. Poole, Y. Hong, A. J. Thompson, G. J. King, J. J. Giovannoni, and G. B. Seymour. 2006. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38: 948–952.
- Martin, A., C. Troadec, A. Boualem, M. Rajab, R. Fernandez, H. Morin, M. Pitrat, C. Dogimont and A. Bendahmane. 2009. A transposon-induced epigenetic change leads to sex determination in melon. Nature 461: 1135–1138.
- Martinez, J. and T. Tuschl. 2004. RISC is a 5′ phosphomonoester-producing RNA endonuclease. Genes Dev. 18: 975–980.
- Mastan, S. G., M. S. Rathore, V. D. Bhatt, P. Yadav and J. Chikara. 2012. Assessment of changes in DNA methylation by methylation-sensitive amplification polymorphism in Jatropha curcas L. subjected to salinity stress. Gene 508: 125–129.
- Matzke, M. A. and R. A. Mosher. 2014. RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15: 394–408.
- Mehraj, H., D. J. Shea, S. Takahashi, N. Miyaji, A. Akter, M. Seki, E. S. Dennis, R. Fujimoto and K. Osabe. 2021b. Genome-wide analysis of long noncoding RNAs, 24-nt siRNAs, DNA methylation and H3K27me3 marks in Brassica rapa. PLoS One 16: e0242530. DOI: 10.1371/journal.pone.0242530.
- Mehraj, H., S. Takahashi, N. Miyaji, A. Akter, Y. Suzuki, M. Seki, E. S. Dennis and R. Fujimoto. 2021a. Characterization of histone H3 Lysine 4 and 36 Tri-methylation in Brassica rapa L. Front. Plant Sci. 12: 659634. DOI: 10.3389/fpls.2021.659634.
- Michalowsky, L. A. and P. A. Jones. 1987. Differential nuclear protein binding to 5-azacytosine-containing DNA as a potential mechanism for 5-aza-2′-deoxycytidine resistance. Mol. Cell Biol. 7: 3076–3083.
- Mitter, N., E. A. Worrall, K. E. Robinson, P. Li, R. G. Jain, C. Taochy, S. J. Fletcher, B. J. Carroll, G. Q. (Max) Lu and Z. P. Xu. 2017a. Clay nanosheets for topical delivery of RNAi for sustained protection against plant viruses. Nat. Plants 3: 16207. DOI: 10.1038/nplants.2016.207.
- Mitter, N., E. A. Worrall, K. E. Robinson, Z. P. Xu and B. J. Carroll. 2017b. Induction of virus resistance by exogenous application of double-stranded RNA. Curr. Opin. Virol. 26: 49–55.
- Miura, K. and T. Furumoto. 2013. Cold signaling and cold response in plants. Int. J. Mol. Sci. 14: 5312–5337.
- Miyaji, N. and R. Fujimoto. 2018. Hybrid vigor: Importance of epigenetic processes and consequences for breeding. Adv. Bot. Res. 88: 247–275.
- Mousavi, S., L. Regni, M. Bocchini, R. Mariotti, N. G. M. Cultrera, S. Mancuso, J. Googlani, M. R. Chakerolhosseini, C. Guerrero, E. Albertini, L. Balondi and P. Proietti. 2019. Physiological, epigenetic and genetic regulation in some olive cultivars under salt stress. Sci. Rep. 9: 1093. DOI: 10.1038/s41598-018-37496-5.
- Murase, K., H. Shiba, M. Iwano, F. S. Che, M. Watanabe, A. Isogai and S. Takayama. 2004. A membrane-anchored protein kinase involved in Brassica self-incompatibility signaling. Science 303: 1516–1519.
- Murase, K., Y. Moriwaki, T. Mori, X. Liu, C. Masaka, Y. Takada, R. Maesaki, M. Mishima, S. Fujii, Y. Hirano, Z. Kawabe, K. Nagata, T. Terada, G. Suzuki, M. Watanabe, K. Shimizu, T. Hakoshima and S. Takayama. 2020. Mechanism of self/nonself-discrimination in Brassica self-incompatibility. Nat. Commun. 11: 4916. DOI: 10.1038/s41467-020-18698-w.
- Niederhuth, C. E. and R. J. Schmitz. 2014. Covering your bases: inheritance of DNA methylation in plant genomes. Mol. Plant 7: 472–480.
- Nou, I. S., M. Watanabe, A. Isogai and K. Hinata. 1993. Comparison of S-alleles and S-glycoproteins between two wild populations of Brassica campestris in Turkey and Japan. Sex. Plant Reprod. 6: 79–86.
- Ohata, M., Y. Takada, Y. Sato, T. Okamoto, K. Murase, S. Takayama, G. Suzuki and M. Watanabe. 2023. MLPK function is not required for self-incompatibility in the S29 haplotype of Brassica rapa L. Plant Reprod. 36: 255–262.
- Ong-Abdullah, M., J. M. Ordway, N. Jiang, S. E. Ooi, S. Y. Kok, N. Sarpan, N. Azimi, A. T. Hashim, Z. Ishak, S. K. Rosli, F. A. Malike, N. A. Bakar, M. Marjuni, N. Abdullah, Z. Yaakub, M. D. Amiruddin, R. Nookiah, R. Singh, E. T. Low, K. L. Chan, N. Azizi, S. W. Smith, B. Bacher, M. A. Budiman, A. V. Brunt, C. Wischmeyer, M. Beil, M. Hogan, N. Lakey, C. C. Lim, X. Arulandoo, C. K. Wong, C. N. Choo, W. C. Wong, Y. Y. Kwan, S. S. Alwee, R. Sambanthamurthi and R. A. Martienssen. 2015. Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature 525: 533–537.
- Papikian, A., W. Liu, J. Gallego-Bartolomé and S. E. Jacobsen. 2019. Site-specific manipulation of Arabidopsis loci using CRISPR-Cas9 SunTag systems. Nat. Commun. 10: 729. DOI: 10.1038/s41467-019-08736-7.
- Park, P. J. 2009. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10: 669–680.
- Parkin, I. A., C. Koh, H. Tang, S. J. Robinson, S. Kagale, W. E. Clarke, C. D. Town, J. Nixon, V. Krishnakumar, S. L. Bidwell, F. Denoeud, H. Belcram, M. G. Links, J. Just, C. Clarke, T. Bender, T. Huebert, A. S. Mason, J. C. Pires, G. Barker, J. Moore, P. G. Walley, S. Manoli, J. Batley, D. Edwards, M. N. Nelson, X. Wang, A. H. Paterson, G. King, I. Bancroft, B. Chalhoub and A. G. Sharpe. 2014. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 15: R77. DOI: 10.1186/gb-2014-15-6-r77.
- Payá-Milans, M., L. Poza-Viejo, P. S. Martín-Uriz, D. Lara-Astiaso, M. D. Wilkinson and P. Crevillén. 2019. Genome-wide analysis of the H3K27me3 epigenome and transcriptome in Brassica rapa. Gigascience 8: giz147. DOI: 10.1093/gigascience/giz147.
- Rodriguez-Granados, N. Y., J. S. Ramirez-Prado, R. Brik-Chaouche, J. An, D. Manza-Mianza, S. Sircar, C. Troadec, M. Hanique, C. Soulard, R. Costa, C. Dogimont, D. Latrasse, C. Raynaud, A. Boualem, M. Benhamed and A. Bendahmane. 2022. CmLHP1 proteins play a key role in plant development and sex determination in melon (Cucumis melo). Plant J. 109: 1213–1228.
- Rodríguez Lorenzo, J. L., M. Hubinský, B. Vyskot and R. Hobza. 2020. Histone post-translational modifications in Silene latifolia X and Y chromosomes suggest a mammal-like dosage compensation system. Plant Sci. 299: 110528. DOI: 10.1016/j.plantsci.2020.110528.
- Rodríguez Lorenzo, J. L., R. Hobza and B. Vyskot. 2018. DNA methylation and genetic degeneration of the Y chromosome in the dioecious plant Silene latifolia. BMC Genomics 19: 540. DOI: 10.1186/s12864-018-4936-y.
- San Miguel, K. and J. G. Scott. 2016. The next generation of insecticides: dsRNA is stable as a foliar-applied insecticide. Pest Manag. Sci. 72: 801–809.
- Sammarco, I., B. Díez Rodríguez, D. Galanti, A. Nunn, C. Becker, O. Bossdorf, Z. Münzbergová and V. Latzel. 2024. DNA methylation in the wild: epigenetic transgenerational inheritance can mediate adaptation in clones of wild strawberry (Fragaria vesca). New Phytol. 241: 1621–1635.
- Schopfer, C. R., M. E. Nasrallah and J. B. Nasrallah. 1999. The male determinant of self-incompatibility in Brassica. Science 286: 1697–1700.
- Schwarz, D. S., G. Hutvágner, T. Du, Z. Xu, N. Aronin and P. D. Zamore. 2003. Asymmetry in the assembly of the RNAi enzyme complex. Cell 115: 199–208.
- Segawa, T., C. Nishiyama, M. Tamiru-Oli, Y. Sugihara, A. Abe, H. Sone, N. Itoh, M. Asukai, A. Uemura, K. Oikawa, H. Utsushi, A. Ikegami-Katayama, T. Imamura, M. Mori, R. Terauchi and H. Takagi. 2021. Sat-BSA: an NGS-based method using local de novo assembly of long reads for rapid identification of genomic structural variations associated with agronomic traits. Breed. Sci. 71: 299–312.
- Shea, D. J., N. Nishida, S. Takada, E. Itabashi, S. Takahashi, A. Akter, N. Miyaji, K. Osabe, H. Mehraj, M. Shimizu, M. Seki, T. Kakizaki, K. Okazaki, E. S. Dennis and R. Fujimoto. 2019. Long noncoding RNAs in Brassica rapa L. following vernalization. Sci. Rep. 9: 9302. DOI: 10.1038/s41598-019-45650-w.
- Shiba, H., M. Iwano, T. Entani, K. Ishimoto, H. Shimosato, F. S. Che, Y. Satta, A. Ito, Y. Takada, M. Watanabe, A. Isogai and S. Takayama. 2002. The dominance of alleles controlling self-incompatibility in Brassica pollen is regulated at the RNA level. Plant Cell 14: 491–504.
- Shimosato, H., N. Yokota, H. Shiba, M. Iwano, T. Entani, F.-S. Che, M. Watanabe, A. Isogai and S. Takayama. 2007. Characterization of the SP11/SCR High-affinity binding site involved in self/nonself recognition in Brassica self-incompatibility. Plant Cell 19: 107–117.
- Shiraki, S., K. Fujiwara, Y. Kamiya, M. A. Akter, E. S. Dennis, R. Fujimoto and H. Mehraj. 2023. Studies on the molecular basis of heterosis in Arabidopsis thaliana and vegetable crops. Horticulturae 9: 366. DOI: 10.3390/horticulturae9030366.
- Stelpflug, S. C., S. R. Eichten, P. J. Hermanson, N. M. Springer and S. M. Kaeppler. 2014. Consistent and heritable alterations of DNA methylation are induced by tissue culture in maize. Genetics 198: 209–218.
- Stone, S. L., M. Arnoldo and D. R. Goring. 1999. A breakdown of Brassica self-incompatibility in ARC1 antisense transgenic plants. Science 286: 1729–1731.
- Stroud, H., B. Ding, S. A. Simon, S. Feng, M. Bellizzi, M. Pellegrini, G. L. Wang, B. C. Meyers and S. E. Jacobsen. 2013. Plants regenerated from tissue culture contain stable epigenome changes in rice. eLife 2: e00354. DOI: 10.7554/eLife.00354.
- Suzuki, G., N. Kai, T. Hirose, K. Fukui, T. Nishio, S. Takayama, A. Isogai, M. Watanabe and K. Hinata. 1999. Genomic organization of the S locus: identification and characterization of genes in SLG/SRK region of S9 haplotype of Brassica campestris (syn. rapa). Genetics 153: 391–400.
- Swain, T., C. Pflueger, S. Freytag, D. Poppe, J. Pflueger, T. V. Nguyen, J. K. Li and R. Lister. 2024. A modular dCas9-based recruitment platform for combinatorial epigenome editing. Nucleic Acids Res. 52: 474–491.
- Takada, S., A. Akter, E. Itabashi, N. Nishida, D. J. Shea, N. Miyaji, H. Mehraj, K. Osabe, M. Shimizu, T. Takasaki-Yasuda, T. Kakizaki, K. Okazaki, E. S. Dennis and R. Fujimoto. 2019. The role of FRIGIDA and FLOWERING LOCUS C genes in flowering time of Brassica rapa leafy vegetables. Sci. Rep. 9: 13843. DOI: 10.1038/s41598-019-50122-2.
- Takahashi, S., K. Osabe, N. Fukushima, S. Takuno, N. Miyaji, M. Shimizu, T. Takasaki-Yasuda, Y. Suzuki, E. S. Dennis, M. Seki and R. Fujimoto. 2018. Genome-wide characterization of DNA methylation, small RNA expression, and histone H3 lysine nine di-methylation in Brassica rapa L. DNA Res. 25: 511–520.
- Takasaki, T., K. Hatakeyama, G. Suzuki, M. Watanabe, A. Isogai and K. Hinata. 2000. The S receptor kinase determines self-incompatibility in Brassica stigma. Nature 403: 913–916.
- Takayama, S., H. Shiba, M. Iwano, H. Shimosato, F. S. Che, N. Kai, M. Watanabe, G. Suzuki, K. Hinata and A. Isogai. 2000. The pollen determinant of self-incompatibility in Brassica campestris. Proc. Natl. Acad. Sci. USA 97: 1920–1925.
- Takayama, S., H. Shimosato, H. Shiba, M. Funato, F. S. Che, M. Watanabe, M. Iwano and A. Isogai. 2001. Direct ligand-receptor complex interaction controls Brassica self-incompatibility. Nature 413: 534–538.
- Tan, C., J. Ren, L. Wang, X. Ye, W. Fu, J. Zhang, M. Qi, H. Feng and Z. Liu. 2021. A single amino acid residue substitution in BraA04g017190.3C, a histone methyltransferase, results in premature bolting in Chinese cabbage (Brassica rapa L. ssp. Pekinensis). BMC Plant Biol. 21: 373. DOI: 10.1186/s12870-021-03153-9.
- Tang, M., W. Xue, X. Li, L. Wang, M. Wang, W. Wang, X. Yin, B. Chen, X. Qu, J. Li, Y. Wu, X. Gao, X. Wei, F. Bu, L. Zhang, Z. Sui, B. Ding, Y. Wang, Q. Zhang, Y. Li and Y. Zhang. 2022. Mitotically heritable epigenetic modifications of CmMYB6 control anthocyanin biosynthesis in chrysanthemum. New Phytol. 236: 1075–1088.
- Tanurdzic, M., M. W. Vaughn, H. Jiang, T. J. Lee, R. K. Slotkin, B. Sosinski, W. F. Thompson, R. W. Doerge and R. A. Martienssen. 2008. Epigenomic consequences of immortalized plant cell suspension culture. PLoS Biol. 6: e302. DOI: 10.1371/journal.pbio.0060302.
- Tarutani, Y., H. Shiba, M. Iwano, T. Kakizaki, G. Suzuki, M. Watanabe, A. Isogai and S. Takayama. 2010. Trans-acting small RNA determines dominance relationships in Brassica self-incompatibility. Nature 466: 983–986.
- Teyssier, E., G. Bernacchia, S. Maury, A. How Kit, L. Stammitti-Bert, D. Rolin and P. Gallusci. 2008. Tissue dependent variations of DNA methylation and endoreduplication levels during tomato fruit development and ripening. Planta 228: 391–399.
- Thompson, K. F. and J. P. Taylor. 1966. Non-linear dominance relationships between S alleles. Heredity 21: 345–362.
- Tirnaz, S. and J. Batley. 2019. Epigenetics: Potentials and challenges in crop breeding. Mol. Plant 12: 1309–1311.
- Tirnaz, S., N. Miyaji, S. Takuno, P. E. Bayer, M. Shimizu, M. A. Akter, D. Edwards, J. Batley and R. Fujimoto. 2022. Whole-genome DNA methylation analysis in Brassica rapa subsp. perviridis in response to Albugo candida infection. Front. Plant Sci. 13: 849358. DOI: 10.3389/fpls.2022.849358.
- Tomari, Y., T. Du, B. Haley, D. S. Schwarz, R. Bennett, H. A. Cook, B. S. Koppetsch, W. E. Theurkauf and P. D. Zamore. 2004. RISC assembly defects in the Drosophila RNAi mutant armitage. Cell 116: 831–841.
- Turcotte, H., J. Hooker, B. Samanfar and J. S. Parent. 2022. Can epigenetics guide the production of better adapted cultivars? Agronomy 12: 838. DOI: 10.3390/agronomy12040838.
- Vaisvila, R., V. K. C. Ponnaluri, Z. Sun, B. W. Langhorst, L. Saleh, S. Guan, N. Dai, M. A. Campbell, B. S. Sexton, K. Marks, M. Samaranayake, J. C. Samuelson, H. E. Church, E. Tamanaha, I. R. Corrêa Jr., S. Pradhan, E. T. Dimalanta, T. C. Evans Jr., L. Williams and T. B. Davis. 2021. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res. 31: 1280–1289.
- Vimont, N., F. X. Quah, D. G. Schöepfer, F. Roudier, E. Dirlewanger, P. A. Wigge, B. Wenden and S. Cortijo. 2020. ChIP-seq and RNA-seq for complex and low-abundance tree buds reveal chromatin and expression co-dynamics during sweet cherry bud dormancy. Tree Genet. Genomes 16: 9. DOI: 10.1007/s11295-019-1395-9.
- Vongs, A., T. Kakutani, R. A. Martienssen and E. J. Richards. 1993. Arabidopsis thaliana DNA methylation mutants. Science 260: 1926–1928.
- Voogd, C., L. A. Brian, R. Wu, T. Wang, A. C. Allan and E. Varkonyi-Gasic. 2022. A MADS-box gene with similarity to FLC is induced by cold and correlated with epigenetic changes to control budbreak in kiwifruit. New Phytol. 233: 2111–2126.
- Wang, Q., Y. Wang, H. Sun, L. Sun and L. Zhang. 2020. Transposon-induced methylation of the RsMYB1 promoter disturbs anthocyanin accumulation in red-fleshed radish. J. Exp. Bot. 71: 2537–2550.
- Wang, S., W. Yan, X. Yang, J. Zhang and Q. Shi. 2021. Comparative methylome reveals regulatory roles of DNA methylation in melon resistance to Podosphaera xanthii. Plant Sci. 309: 110954. DOI: 10.1016/j.plantsci.2021.110954.
- Wang, Z. and D. C. Baulcombe. 2020. Transposon age and non-CG methylation. Nat. Commun. 11: 1221. DOI: 10.1038/s41467-020-14995-6.
- Wang, Z., D. Meng, A. Wang, T. Li, S. Jiang, P. Cong and T. Li. 2013. The methylation of the PcMYB10 promoter is associated with green-skinned sport in Max Red Bartlett pear. Plant Physiol. 162: 885–896.
- Wassenegger, M., S. Heimes, L. Riedel and H. L. Sänger. 1994. RNA-directed de novo methylation of genomic sequences in plants. Cell 76: 567–576.
- Whittaker, C. and C. Dean. 2017. The FLC locus: a platform for discoveries in epigenetics and adaptation. Ann. Rev. Cell Dev. Biol. 33: 555–575.
- Wu, Q., Q. Huang, H. Guan, X. Zhang, M. Bao, M. Bendahmane and X. Fu. 2022. Comprehensive genome-wide analysis of histone acetylation genes in roses and expression analyses in response to heat stress. Genes (Basel) 13: 980. DOI: 10.3390/genes13060980.
- Xi, Y., S. R. Park, D. H. Kim, E. D. Kim and S. Sung. 2020. Transcriptome and epigenome analyses of vernalization in Arabidopsis thaliana. Plant J. 103: 1490–1502.
- Xie, L., X. Bai, H. Zhang, X. Qiu, H. Jian, Q. Wang, H. Wang, D. Feng, K. Tang and H. Yan. 2023. Loss of rose fragrance under chilling stress is associated with changes in DNA methylation and volatile biosynthesis. Genes (Basel) 14: 692. DOI: 10.3390/genes14030692.
- Xu, J., G. Chen, P. J. Hermanson, Q. Xu, C. Sun, W. Chen, Q. Kan, M. Li, P. A. Crisp, J. Yan, L. Li, N. M. Springer and Q. Li. 2019. Population-level analysis reveals the widespread occurrence and phenotypic consequence of DNA methylation variation not tagged by genetic variation in maize. Genome Biol. 20: 243. DOI: 10.1186/s13059-019-1859-0.
- Xu, J., S. Zhou, X. Gong, Y. Song, S. van Nocker, F. Ma and Q. Guan. 2018. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 16: 672–687.
- Yamamoto, M., T. Ishii, M. Ogura, T. Akanuma, X. Y. Zhu and H. Kitashiba. 2023. S haplotype collection in Brassicaceae crops—an updated list of S haplotypes. Breed. Sci. 73: 132–145.
- Yang, X., H. Kundariya, Y. Z. Xu, A. Sandhu, J. Yu, S. F. Hutton, M. Zhang and S. A. Mackenzie. 2015. MutS HOMOLOG1-derived epigenetic breeding potential in tomato. Plant Physiol. 168: 222–232.
- Yao, Z., L. Yuan, K. Liu, T. Wang, B. Liu, Y. Zhao, S. Gan and L. Chen. 2022. Warming-induced changes of broccoli head to cauliflower-like curd in Brassica oleracea are regulated by DNA methylation as revealed by methylome and transcriptome co-profiling. Mol. Hortic. 2: 26. DOI: 10.1186/s43897-022-00047-8.
- Yasuda, S., R. Kobayashi, T Ito, Y. Wada and S. Takayama. 2021. Homology-based interactions between small RNAs and their targets control dominance hierarchy of male determinant alleles of self-incompatibility in Arabidopsis lyrata. Int. J. Mol. Sci. 22: 6990. DOI: 10.3390/ijms22136990.
- Yasuda, S., Y. Wada, T. Kakizaki, Y. Tarutani, E. Miura-Uno, K. Murase, S. Fujii, T. Hioki, T. Shimoda, Y. Takada, H. Shiba, T. Takasaki-Yasuda, G. Suzuki, M. Watanabe and S. Takayama. 2016. A complex dominance hierarchy is controlled by polymorphism of small RNAs and their targets. Nat. Plants 3: 16206.
- Yu, L., Y. Sun, X. Zhang, M. Chen, T. Wu, J. Zhang, Y. Xing, J. Tian and Y. Yao. 2022. ROS1 promotes low temperature-induced anthocyanin accumulation in apple by demethylating the promoter of anthocyanin-associated genes. Hortic. Res. 9: uhac007. DOI: 10.1093/hr/uhac007.
- Zeng, Z., W. Zhang, A. P. Marand, B. Zhu, C. R. Buell and J. Jiang. 2019. Cold stress induces enhanced chromatin accessibility and bivalent histone modifications H3K4me3 and H3K27me3 of active genes in potato. Genome Biol. 20: 123. DOI: 10.1186/s13059-019-1731-2.
- Zhang, H., Z. Lang and J. K. Zhu. 2018. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 19: 489–506.
- Zhang, L., J. Huang, S. Su, X. Wei, L. Yang, H. Zhao, J. Yu, J. Wang, J. Hui, S. Hao, S. Song, Y. Cao, M. Wang, X. Zhang, Y. Zhao, Z. Wang, W. Zheng, H. M. Wu, Y. Yuan, X. Zhang, A. Y. Cheung and Q. Duan. 2021. FERONIA receptor kinase-regulated reactive oxygen species mediate self-incompatibility in Brassica rapa. Curr. Biol. 31: 3004–3016.
- Zhang, Q., Y. Li, T. Xu, A. K. Srivastava, D. Wang, L. Zeng, L. Yang, L. He, H. Zhang, Z. Zheng, D. L. Yang, C. Zhao, J. Dong, Z. Gong, R. Liu and J. K. Zhu. 2016. The chromatin remodeler DDM1 promotes hybrid vigor by regulating salicylic acid metabolism. Cell Discov. 2: 16027. DOI: 10.1038/celldisc.2016.27.
- Zhang, W., X. Wang, Q. Yu, R. Ming and J. Jiang. 2008. DNA methylation and heterochromatinization in the male-specific region of the primitive Y chromosome of papaya. Genome Res. 18: 1938–1943.
- Zhang, Y., J. Zhong, A. Munawar, Y. Cai, W. He, Y. Zhang, H. Guo, Y. Gao, Z. Zhu and W. Zhou. 2024. Knocking down a DNA demethylase gene affects potato plant defense against a specialist insect herbivore. J. Exp. Bot. 75: 483–499.
- Zhao, Y., Y. Li, K. Cao, W. Fang, C. Chen, X. Wang, J. Wu, W. Guo and L. Wang. 2024. Peculiarity of transcriptional and H3K27me3 dynamics during peach bud dormancy. Hortic. Plant J. 10: 38–50.
- Zhong, S., Z. Fei, Y. R. Chen, Y. Zheng, M. Huang, J. Vrebalov, R. McQuinn, N. Gapper, B. Liu, J. Xiang, Y. Shao and J. J. Giovannoni. 2013. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 31: 154–159.
- Zhou, L., S. Tian and G. Qin. 2019. RNA methylomes reveal the m6A-mediated regulation of DNA demethylase gene SlDML2 in tomato fruit ripening. Genome Biol. 20: 156. DOI: 10.1186/s13059-019-1771-7.
- Zhu, H., P. Y. Chen, S. Zhong, C. Dardick, A. Callahan, Y. Q. An, S. van Knocker, Y. Yang, G. Y. Zhong, A. Abbott and Z. Liu. 2020a. Thermal-responsive genetic and epigenetic regulation of DAM cluster controlling dormancy and chilling requirement in peach floral buds. Hortic. Res. 7: 114. DOI: 10.1038/s41438-020-0336-y.
- Zhu, Y. C., B. Zhang, A. C. Allan, K. Lin-Wang, Y. Zhao, K. Wang, K. S. Chen and C. J. Xu. 2020b. DNA demethylation is involved in the regulation of temperature-dependent anthocyanin accumulation in peach. Plant J. 102: 965–976.
