Abstract
During the evolution of skeletons, terrestrial vertebrates acquired strong bones made of calcium–phosphate. By keeping the extracellular fluid in a supersaturated condition regarding calcium and phosphate ions, they created the bone when and where they wanted simply by providing a cue for precipitation. To secure this strategy, they acquired a novel endocrine system to strictly control the extracellular phosphate concentration. In response to phosphate intake, fibroblast growth factor–23 (FGF23) is secreted from the bone and acts on the kidney through binding to its receptor Klotho to increase urinary phosphate excretion, thereby maintaining phosphate homeostasis. The FGF23–Klotho endocrine system, when disrupted in mice, results in hyperphosphatemia and vascular calcification. Besides, mice lacking Klotho or FGF23 suffer from complex aging-like phenotypes, which are alleviated by placing them on a low- phosphate diet, indicating that phosphate is primarily responsible for the accelerated aging. Phosphate acquires the ability to induce cell damage and inflammation when precipitated with calcium. In the blood, calcium–phosphate crystals are adsorbed by serum protein fetuin-A and prevented from growing into large precipitates. Consequently, nanoparticles that comprised calcium–phosphate crystals and fetuin-A, termed calciprotein particles (CPPs), are generated and dispersed as colloids. CPPs increase in the blood with an increase in serum phosphate and age. Circulating CPP levels correlate positively with vascular stiffness and chronic non-infectious inflammation, raising the possibility that CPPs may be an endogenous pro-aging factor. Terrestrial vertebrates with the bone made of calcium– phosphate may be destined to age due to calcium–phosphate in the blood.
Introduction
In 1997, we reported a mouse mutant strain exhibiting a premature aging syndrome
1)
. The mutant was one of the 28 transgenic mouse strains carrying the rabbit type-I sodium–proton exchanger (NHE1) gene
2)
. Only three of them expressed the exogenous rabbit NHE1 gene, whereas the remaining 25 strains did not. Each strain that did not express the transgene was maintained individually to generate mice homozygous for the transgene-inserted allele. One of these strains exhibited complex phenotypes resembling human aging when homozygous for the transgene, suggesting that the insertional mutation of the transgene might have disrupted a putative aging suppressor gene. The gene was named klotho after one of the goddesses of destiny in Greek myth who spins the thread of life. Mice homozygous for the transgene (kl/kl mice) were born in the expected Mendelian ratio and developed normally until weaning. After weaning, they start exhibiting aging-like phenotypes, including growth arrest, atrophy of multiple organs (gonads, thymus, and skin), sarcopenia, reduced bone mineral density
3)
, cardiac hypertrophy
4)
, vascular calcification, emphysematous lung, hearing disturbance
5)
, cognition impairment
6)
, frailty, and premature death approximately 2 months of age. The klotho gene was identified by positional cloning using the transgene as a marker and turned out to encode a novel single-pass transmembrane protein expressed predominantly in renal tubular cells
1)
. As it was a new gene, there was no clue to know its function. It took almost 10 years before the function of Klotho protein became evident.
The FGF23–Klotho Endocrine Axis
In 2004, mice lacking fibroblast growth factor–23 (FGF23) were reported
7)
. At that time, it had been known that patients carrying gain-of-function mutations in the FGF23 gene
8)
or tumors producing FGF23
9)
exhibited hypophosphatemia associated with phosphate wasting into the urine, disturbed bone mineralization (osteomalacia or rickets), and inappropriately low serum levels of active vitamin D (1,25-dihydroxyvitamin D3). Hence, FGF23 was regarded as a hormone that promoted urinary phosphate excretion and counter-regulated active vitamin D
10)
. Expectedly, mice lacking FGF23 showed high serum phosphate and active vitamin D levels associated with vascular calcification. Besides these predictable phenotypes, these mice exhibited growth retardation, atrophy of gonads and thymus, reduced bone mineral, and shortened life span. Because mice lacking Klotho also had high serum phosphate and active vitamin D levels
11)
, we realized that FGF23 knockout mice were a phenocopy of Klotho- deficient mice. Prompted by this observation, we pursued a potential link between FGF23 and Klotho and found that Klotho functioned as a receptor for FGF23
12)
. More precisely, Klotho protein forms constitutive binary complexes with specific isoforms of fibroblast growth factor receptor (FGFR1c, FGFR3c, and FGFR4). Although FGF23 has very low affinity to these FGFR isoforms and barely binds to them at its physiological concentration
13)
, it can bind to the Klotho–FGFR complexes with high affinity, indicating that Klotho functions as the obligate co-receptor for FGF23
12)
. Nine months later, another laboratory published a paper that reproduced the same finding
14)
. The fact that FGF23 requires Klotho to exert its function explains the striking similarity in phenotypes between FGF23 knockout mice and Klotho-deficient mice.
These seminal studies identified the FGF23–Klotho system as a novel endocrine axis indispensable for maintaining phosphate homeostasis
15)
. In response to phosphate intake, FGF23 is secreted from the bone (osteocytes and osteoblasts) and binds to the FGFR–Klotho complexes expressed on renal tubular cells to suppress phosphate reabsorption and increase urinary phosphate excretion, thereby balancing between intake and excretion of phosphate. FGF23 also has the activity that inhibits the synthesis and promotes the degradation of active vitamin D in renal tubules, thereby suppressing calcium absorption from the intestine to prevent an increase in circulating levels of calciprotein particles (CPPs), which will be discussed later.
In 2018, the crystal structure of the Klotho protein was solved (
Fig. 1)
16
,
17)
. Similar to its namesake in Greek myth, Klotho protein sends out a long “thread” with an intrinsically disordered structure, which is termed the receptor binding arm (RBA) because it directly interacts with FGFR. Once the RBA captures FGFR and takes a solid structure, a high-affinity binding pocket for FGF23 is generated between Klotho and FGFR. The formation of the FGF23–FGFR–Klotho ternary complex is required to activate FGFR tyrosine kinase and the canonical intracellular FGF signaling pathway.
Phosphate Accelerates Aging
Disruption of either FGF23 or Klotho leads to phosphate retention and elevation of serum active vitamin D levels
1,
7,
11)
. Because active vitamin D promotes calcium absorption from the intestine, hypercalcemia ensues. The question is which was responsible for the complex aging-like phenotypes in Klotho-deficient mice and FGF23- deficient mice, phosphate, active vitamin D, or calcium. Several interventions that lowered blood phosphate, calcium, and/or active vitamin D levels were reported to alleviate the aging-like phenotypes in these mice, including feeding of low-phosphate diet
18,
19)
or vitamin D deficient diet
11,
19)
, ablation of the gene encoding vitamin D receptor
20)
, 1α-hydroxylase
21)
, or sodium–phosphate co-transporter type IIa (Npt2a)
22)
. 1α-hydroxylase is an enzyme expressed in renal proximal tubules and required for the synthesis of active vitamin D. Npt2a is expressed on the apical membrane of renal proximal tubules and responsible for phosphate reabsorption. These dietary and genetic interventions consistently lowered serum phosphate levels, whereas the changes in serum calcium and active vitamin D levels varied (
Table 1)
. Notably, when mice lacking FGF23 were placed on a low-phosphate diet, their serum levels of active vitamin D were further elevated, which was a physiological response to the low-phosphate diet. Despite that, the low-phosphate diet alleviated their aging-like phenotypes
19)
, indicating that phosphate, but not active vitamin D or calcium, was the true culprit of accelerated aging in mice lacking FGF23 or Klotho. In other words, phosphate accelerated aging.
Table 1.
Interventions that alleviate aging-like phenotypes in mice lacking FGF23 and/or Klotho and associated changes in serum levels of phosphate, calcium, and 1,25-dihydroxyvitamin D
3 (vitamin D
59)
(modified from Ref.59)
| Interventions |
Changes in serum levels |
| Phosphate |
Calcium |
Vitamin D |
| Vitamin D-deficient diet |
↓ |
↓ |
↓ |
| Ablation of vitamin D receptor |
↓ |
↓ |
↓ |
| Ablation of 1α-hydroxylase |
↓ |
↓ |
↓ |
| Ablation of Npt2a |
↓ |
↑ |
↑ |
| Low-phosphate diet |
↓ |
↑ |
↑ |
There are several lines of circumstantial evidence in support of this notion. Serum phosphate levels inversely correlate with average life spans in mammals (
Fig. 2)
23)
. In humans, high serum phosphate levels are associated with high all-cause mortality
24)
. Then, what is the mechanism by which phosphate accelerates aging?
Evolution of the Bone Made of Calcium Phosphate
Consistent with the notion that life is derived from the sea, the composition of the seawater and the human body is similar. Indeed, nine out of the top 10 abundant elements are identical between them (
Table 2)
25)
. The exceptions are magnesium in seawater and phosphorus in the human body, suggesting that organisms have started accumulating phosphorus at some point during evolution. Accumulation of phosphorus occurred about 400 million years ago in the Devonian era when the bony fish evolved
25,
26)
. Organisms after the bony fish accumulate a large amount of phosphorus in the bone in the form of calcium–phosphate. Organisms before the bony fish have skeletons made of calcium–carbonate or cartilage. Because the bone made of calcium–phosphate is physically harder than the skeletons made of calcium–carbonate or cartilage, acquisition of the strong bone made of calcium– phosphate may be a prerequisite for the evolution of terrestrial vertebrates that are required to support their own body weight and move around without the help of water buoyancy. To create the bone made of calcium–phosphate, terrestrial vertebrates maintain the extracellular fluid in a supersaturated condition regarding calcium and phosphate ions and provide a cue for precipitation when and where they want to make the bone. To secure this strategy, they acquired two major mechanisms. First, the FGF23–Klotho endocrine system has evolved to strictly control the extracellular phosphate concentration. Klotho orthologs exist only in organisms that have bones made of calcium– phosphate
26)
. Second, they have developed defense mechanisms to prevent the growth of calcium phosphate precipitation in extraosseous tissues. One of such defense mechanisms is the formation of CPPs.
Table 2.
The top 10 abundant elements in seawater and the human body
25,
60)
. The water abundance (by the number of H and O atoms) in seawater was reduced by boiling to the level equivalent to that in humans (modified from Ref. 25)
|
Seawater |
Human body |
| 1 |
H |
H |
| 2 |
O |
O |
| 3 |
Na |
C |
| 4 |
Cl |
N |
| 5 |
Mg |
Na |
| 6 |
S |
Ca |
| 7 |
K |
P |
| 8 |
Ca |
S |
| 9 |
C |
K |
| 10 |
N |
Cl |
Physiology of CPPs
Because blood is a supersaturated solution in terms of calcium and phosphate ions, even a slight and transient increase in the blood phosphate concentration after meal (postprandial hyperphosphatemia) can trigger precipitation of tiny amorphous calcium–phosphate (
Fig. 3)
. Such calcium–phosphate precipitates in the blood are immediately adsorbed by serum protein fetuin-A and prevented from growing into large deposits
27
,
28)
. Consequently, fetuin-A molecules laden with amorphous calcium–phosphate are generated, which are termed calciprotein monomers (CPMs) or primary CPPs (aggregates of CPMs). Calcium–phosphate in primary CPPs spontaneously undergo phase transition from the amorphous phase to the crystalline phase to become secondary CPPs over time. These CPPs extravasate through sinusoids in the liver and the bone marrow to be phagocytosed by the reticuloendothelial system and deposited to the bone, respectively
29,
30)
. Thus, CPPs may not merely be a by-product of the defense mechanism that prevents the growth of calcium–phosphate precipitates in the blood but function as a vehicle that carries calcium and phosphate absorbed from the intestine to the bone. Indeed, blood CPP levels were increased within 2 h after phosphate ingestion in mice and then restored to the baseline as CPPs accumulated on the bone surface
30)
.

Additionally, CPPs function as a regulator of the FGF23–Klotho endocrine axis
30)
. FGF23 is secreted from osteocytes and osteoblasts in response to phosphate intake; however, the mechanism by which these cells sense phosphate intake had been elusive. It had been speculated that osteocytes and osteoblasts might sense a postprandial increase in the blood phosphate level through a putative “phosphate-sensing” receptor and secrete FGF23 to lower the blood phosphate level. This hypothesis was prompted by the fact that parathyroid cells sense a decrease in the blood calcium level through the calcium-sensing receptor and secrete parathyroid hormone to raise the blood calcium level
31)
. However, such phosphate-sensing receptor has not been identified yet. Furthermore, some studies challenged this hypothesis. First, serum FGF23 levels correlated not only with serum phosphate but also with serum calcium levels. Second, the increase in serum phosphate failed to increase FGF23 in the presence of hypocalcemia in rodents. Conversely, the increase in serum calcium failed to increase FGF23 in the presence of hypophosphatemia, indicating that both serum phosphate and calcium must be above certain levels to induce FGF23
32,
33)
. These observations can be explained by the fact that CPPs induce FGF23 expression and secretion in osteoblasts
30)
. Consequently, FGF23 promotes urinary phosphate excretion and reduces serum levels of active vitamin D, which lowers blood phosphate levels and suppresses calcium absorption from the intestine (
Fig. 4)
. Therefore, the ability of CPPs to induce FGF23 expression and secretion may be required for a negative feedback loop to prevent an increase in blood CPPs, which can increase the risk for vascular calcification and chronic non- infectious inflammation as will be discussed in the next section.

Pathology of CPPs
CPPs have been reported to exert adverse effects on various types of cultured cells. Secondary CPPs, but not CPMs and primary CPPs, induce calcification and innate immune responses when applied to cultured vascular smooth muscle cells (VSMCs) and macrophages, respectively
34,
35)
. Clinical studies have shown that serum levels of CPPs correlate with parameters of vascular calcification (coronary artery calcification scores), vascular stiffness (pulse wave velocity), and inflammation (hs-CRP) in patients with chronic kidney disease (CKD)
36,
37)
.
In these studies, serum CPP levels were determined by the “fetuin-A method.” First, the fetuin-A level in the serum sample was measured using enzyme-linked immunosorbent assay (ELISA, X mg/mL). Second, the remaining serum sample was centrifuged at 16,000–24,000 g for 2 h to precipitate CPPs. Lastly, the fetuin-A level in the supernatant was measured using ELISA (Y mg/mL). The reduction ratio of the fetuin-A level by the centrifugation, (X–Y)/X was defined as a surrogate of the serum CPP level. This assay is based on the fact that fetuin-A is the major protein component of CPPs. However, the fetuin-A method entails two fundamental limitations. First, the assay becomes increasingly inaccurate as the reduction ratio becomes lower and closer to the coefficient of variation of the ELISA. Second, low-density CPPs that were not precipitated by the centrifugation were not measured.
To overcome these limitations of the fetuin-A method, we developed a novel assay using OsteoSense, an infrared fluorescent probe that specifically binds to calcium–phosphate crystals
38)
. Serum/plasma samples were inoculated with OsteoSense and incubated for an hour to let OsteoSense bind to calcium–phosphate crystals in CPPs. The mixture was then subjected to a gel filtration spin column to remove unbound OsteoSense. The fluorescence intensity of the flow-through fraction quantified by an infrared scanner represented the amount of CPPs. We measured plasma CPP levels in 148 patients with CKD and observed an increase in CPP levels with age, the progression of CKD stages, and an increase in serum phosphate levels. Multivariate analysis identified the serum phosphate level and the age as two independent determinants of the plasma CPP level
38)
.
Notably, chronic, low-grade, non-infectious inflammation is associated with accelerated aging and age-related disorders. This phenomenon is termed the “inflammaging” and regarded as a significant risk for morbidity and mortality in the aged
39,
40)
. In mice, chronic inflammation caused by constitutive activation of the NFκB pathway was reported to accelerate aging
41)
. Specifically, targeted disruption of the Nfκb1 gene encoding p105 subunit of NFκB that functions as a repressor of pro-inflammatory gene transcription induced inflammation in the absence of overt infection and reduced regeneration of liver and intestine, associated with accumulation of senescent cells and shortened life span. Although inflammation has been established as a potent accelerator of aging, endogenous “pathogens” that can trigger non-infectious inflammation responsible for accelerated aging remain to be identified. Damage-associated molecular patterns (DAMPs) are candidates of such pathogens, including ATP, mitochondrial and genomic DNA, RNA, and histones among others
42)
. Given the ability of CPPs to induce cell damage and innate immune responses, CPPs may function as a pathogen of aging.
Mechanism of Vascular Calcification Induced by CPPs
Secondary CPPs, but not primary CPPs, have the activity that induces calcification in cultured VSMCs
34)
. Secondary CPPs activate NFκB signaling in VSMCs to induce phenotypic transformation to osteoblastic cells
43)
. The transformed VSMCs start secreting bone matrix proteins, providing an extracellular scaffold for the deposition of calcium–phosphate. Reportedly, zinc inhibited calcification not only in cultured VSMCs but also in the aorta of Klotho-deficient mice, subtotal nephrectomized mice, and cholecalciferol-overloaded mice
44)
. Zinc binds to a “zinc- sensing receptor” GPR39 expressed on the surface of VSMCs and induces expression of a zinc-finger protein TNFα-induced protein 3, which functions as a potent inhibitor of NFκB activation (
Fig. 5)
. Therefore, zinc suppresses vascular calcification primarily through inhibiting the osteoblastic transformation of VSMCs.
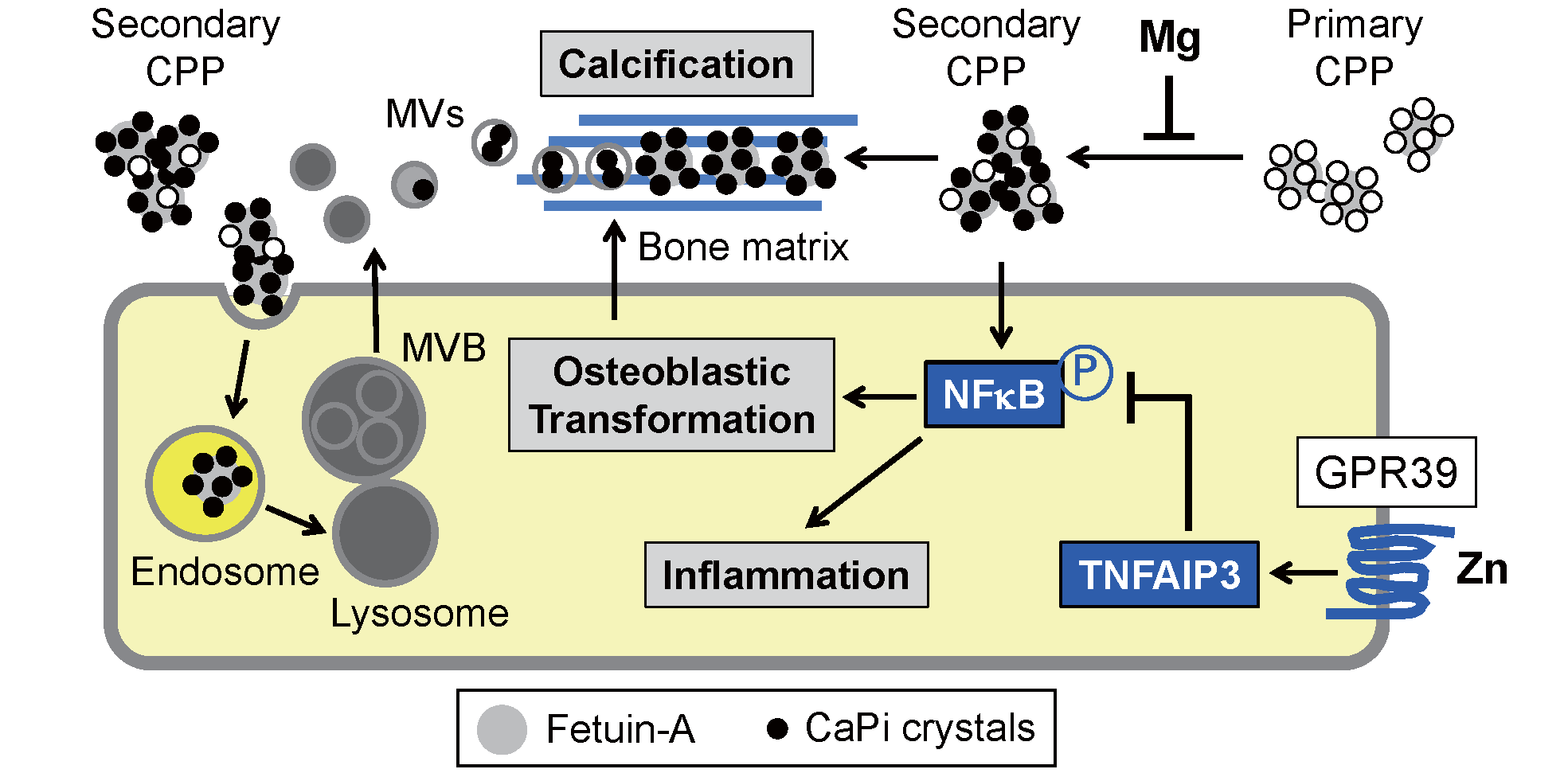
Magnesium can also inhibit vascular calcification, as evidenced by the fact that a high magnesium diet rescued Klotho-deficient mice from aortic calcification
45)
. Furthermore, a randomized control trial demonstrated that the administration of magnesium oxide suppressed the progression of coronary artery calcification in patients with stages 3 and 4 CKD
46)
. The beneficial effect of magnesium on vascular calcification may depend on its ability to suppress the transition of primary CPPs to secondary CPPs (
Fig. 5)
47)
.
How CPPs fuel calcium–phosphate to calcified lesions remains to be determined. Two mechanisms are considered, which are not mutually exclusive. One possible mechanism is that CPPs should be endocytosed by VSMCs to induce vascular calcification (
Fig. 5)
. Although the mode of endocytosis and endosomal pathways through which CPPs are processed in VSMCs remain elusive, inorganic calcium–phosphate nanocrystals were reported to be endocytosed and transferred to lysosomes
48)
, where calcium–phosphate in CPPs can be dissolved under the acidic condition. Hence, the concentration of calcium and phosphate in the lysosomes is assumed to be rapidly increased, which potentially induces disruption of lysosomal integrity, leakage of calcium and phosphate to the cytoplasm, and cell death. Indeed, VSMC death induced by calcium–phosphate nanocrystals was interfered with a lysosomal proton pump inhibitor bafilomycin A1 that increases pH in lysosomes
48)
. Given that CPPs are processed similarly as calcium–phosphate nanocrystals by VSMCs, endocytosed CPPs are transferred to lysosomes to induce cell death. Alternatively, the lysosomes laden with CPPs may fuse with multivesicular bodies (MVBs)
49)
to provide them with calcium, phosphate, and fetuin-A. Vesicles within the MVBs are secreted as exosomes containing calcium–phosphate crystals, which are termed matrix vesicles (MVs). Indeed, MVs secreted by calcifying VSMCs are specifically loaded with calcium, phosphate, and fetuin-A and serve as the first nidus for calcification
50)
.
The other mechanism is that CPPs may be deposited directly to the bone matrix without being endocytosed by transformed VSMCs (
Fig. 5)
. This possibility is supported by the fact that CPPs can induce calcification in cultured VSMCs even after they are fixed with formalin or methanol
51)
. In either case, the phenotypic transformation of VSMCs to osteoblastic cells and secretion of bone matrix proteins appear to be a prerequisite for vascular calcification.
Colloids and Arteriosclerosis
Generally, colloids are used as vehicles that transport insoluble materials between organs through the bloodstream. Lipids are one of the major insoluble biological materials. Lipids form complexes with apoproteins to become lipoproteins, which exist as a polydisperse colloidal system that comprised nanoparticles with different physical properties such as size and density
52)
. When lipids, which should be stored in adipose tissues, are deposited to arterial walls, atherosclerosis ensues. (Patho)physiological activity of lipoproteins depends on their physical property; low-density lipoproteins, whose diameter and density are ~25 nm and <1.063 g/mL, respectively, are pro-atherogenic and therefore regarded as “bad” cholesterol. By contrast, high-density lipoproteins, whose diameter and density are ~9 nm and > 1.063 g/mL, respectively, are anti-atherogenic and hence designated as “good” cholesterol
27)
.
Besides lipids, calcium–phosphate is another major insoluble material in mammals. calcium–phosphate forms complexes with fetuin-A to become CPPs with different size, density, and the phase (amorphous or crystalline) of calcium–phosphate
27)
. When calcium–phosphate, which should be stored in the bone, are deposited to arterial walls, vascular calcification ensues. Similar to lipoproteins, the activity of CPPs depends on their physical property. CPMs, which are small (~9 nm in diameter), low in density (not precipitated by centrifugation at 16,000–24,000 g for 2 h), and contain amorphous calcium–phosphate but not crystalline calcium–phosphate, have the potent ability to induce FGF23 expression in osteoblasts and never induce cell damage. Therefore, CPMs are regarded as “good” CPPs. By contrast, secondary CPPs are large (> 35 nm in diameter), high in density (precipitated by centrifugation at 16,000–24,000 g for 2 h), and contain crystalline calcium–phosphate. Secondary CPPs are potent inducers of cell damage, calcification, and innate immune responses
34,
35)
and thus regarded as “bad” CPPs.
Although atherosclerosis and vascular calcification represent two distinct pathologies of arteriosclerosis, they can be sublated as a disorder caused by the mistargeting of colloidal particles in the blood to arteries (
Fig. 6)
15)
.
Hemodialysis Patients and Klotho Deficient Mice
Klotho-deficient mice suffer from hyperphosphatemia. In humans, hyperphosphatemia is universally observed in patients with end-stage renal disease receiving hemodialysis. Because blood phosphate levels positively correlate with blood CPP levels
38)
, both hemodialysis patients and Klotho-deficient mice have high blood CPP levels. Additionally, they share similar aging-like symptoms and pathophysiology, including shortened life span, vascular calcification, cardiac hypertrophy, sarcopenia, frailty, loss of Klotho expression in the kidney, and chronic non- infectious inflammation
53)
. Furthermore, both of them benefit from dietary phosphate restriction. However, Klotho-deficient mice are distinct from hemodialysis patients in that they never suffer from renal failure. Indeed, their serum creatinine levels are not increased
1)
. Hyperphosphatemia in Klotho-deficient mice is caused by an isolated deficiency of urinary phosphate excretion due to refractoriness to FGF23. By contrast, hyperphosphatemia in hemodialysis patients is caused by a generalized loss of renal function. Despite this fundamental difference, hemodialysis patients and Klotho-deficient mice show striking similarity in their symptoms, pathophysiology, and response to the treatment. The complex symptoms in hemodialysis patients are collectively designated as uremia and have been believed to be caused by the accumulation of multiple uremic toxins. However, the fact that the isolated deficiency of urinary phosphate excretion leads to pathophysiology closely resembling uremia suggests that phosphate, or more specifically CPPs, may be the most important uremic toxin.
Aging Research in the Future
Basic aging research has focused on understanding mechanisms of accelerated aging and extended longevity that are evolutionarily conserved throughout various species across the animal kingdom, including yeast, nematodes, flies, rodents, and primates. During this research endeavor, several important discoveries have been made including, but not limited to, the following: appropriate inhibition of insulin-like activity
54)
and restriction of calorie intake
55)
extend life span, and the accumulation of senescent cells accelerates organismal aging
56)
. Based on these findings, calorie restriction mimetics and senolytic drugs that selectively kill senescent cells have been
developed, aiming at suppressing aging to solve healthcare problems in the aging society
57,
58)
. By contrast, mechanisms of aging that are specific to particular species have never been explored. However, the serendipitous discovery of the kl/kl mouse exhibiting a premature aging syndrome has revealed that the FGF23–Klotho endocrine system, which is specific to vertebrates with the bone made of calcium–phosphate, leads to phenotypes resembling aging when disrupted
1,
7)
. Furthermore, the fact that phosphate is responsible for the aging-like phenotypes in mice lacking FGF23 or Klotho suggests that phosphate, or more precisely CPPs, may be a pro-aging factor specific to vertebrates. That is, vertebrates with the bone made of calcium–phosphate may be destined to age because of ectopic calcium–phosphate. Studies on the aging mechanism specific to higher animals may open a new avenue for the development of effective anti-aging medicine.
Conflict of Interest
The author received honoraria and research funds from Bayer and Kissei Pharmaceutical.
References
- 1) Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R and Nabeshima Y: Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature, 1997; 390: 45-51
- 2) Kuro-o M, Hanaoka K, Hiroi Y, Noguchi T, Fujimori Y, Takewaki S, Hayasaka M, Katoh H, Miyagishi A, Nagai R and Nabeshima Y: Salt-sensitive hypertension in transgenic mice overexpressing Na(+)-proton exchanger. Circ Res, 1995; 76: 148-153
- 3) Kawaguchi H, Manabe N, Miyaura C, Chikuda H, Nakamura K and Kuro-o M: Independent impairment of osteoblast and osteoclast differentiation in klotho mouse exhibiting low-turnover osteopenia. J Clin Invest, 1999; 104: 229-237
- 4) Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutierrez OM, Aguillon-Prada R, Lincoln J, Hare JM, Mundel P, Morales A, Scialla J, Fischer M, Soliman EZ, Chen J, Go AS, Rosas SE, Nessel L, Townsend RR, Feldman HI, St John Sutton M, Ojo A, Gadegbeku C, Di Marco GS, Reuter S, Kentrup D, Tiemann K, Brand M, Hill JA, Moe OW, Kuro-o M, Kusek JW, Keane MG and Wolf M: FGF23 induces left ventricular hypertrophy. J Clin Invest, 2011; 121: 4393-4408
- 5) Kamemori M, Ohyama Y, Kurabayashi M, Takahashi K, Nagai R and Furuya N: Expression of Klotho protein in the inner ear. Hear Res, 2002; 171: 103-110
- 6) Nagai T, Yamada K, Kim HC, Kim YS, Noda Y, Imura A, Nabeshima Y and Nabeshima T: Cognition impairment in the genetic model of aging klotho gene mutant mice: a role of oxidative stress. Faseb J, 2003; 17: 50-52
- 7) Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K and Yamashita T: Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest, 2004; 113: 561-568
- 8) White KE, Evans WE, O’Rlordan JLH, Speer MC, Econs MJ, Lorenz-Deplereux B, Grabowski M, Meitinger T and Storm TM: Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet, 2000; 26: 345-348
- 9) Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S and Yamashita T: Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A, 2001; 98: 6500-6505
- 10) Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S and Yamashita T: FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res, 2004; 19: 429-435
- 11) Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K and Nabeshima Y: Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol, 2003; 17: 2393-2403
- 12) Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW and Kuro-o M: Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem, 2006; 281: 6120-6123
- 13) Yu X, Ibrahimi OA, Goetz R, Zhang F, Davis SI, Garringer HJ, Linhardt RJ, Ornitz DM, Mohammadi M and White KE: Analysis of the biochemical mechanisms for the endocrine actions of fibroblast growth factor-23. Endocrinology, 2005; 146: 4647-4656
- 14) Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S and Yamashita T: Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature, 2006; 444: 770-774
- 15) Kuro-o M: The Klotho proteins in health and disease. Nat Rev Nephrol, 2019; 15: 27-44
- 16) Chen G, Liu Y, Goetz R, Fu L, Jayaraman S, Hu MC, Moe OW, Liang G, Li X and Mohammadi M: alpha-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature, 2018; 553: 461-466
- 17) Kuro-o M: Ageing-related receptors resolved. Nature, 2018; 553: 409-410
- 18) Morishita K, Shirai A, Kubota M, Katakura Y, Nabeshima Y, Takeshige K and Kamiya T: The progression of aging in klotho mutant mice can be modified by dietary phosphorus and zinc. J Nutr, 2001; 131: 3182-3188
- 19) Stubbs JR, Liu S, Tang W, Zhou J, Wang Y, Yao X and Quarles LD: Role of Hyperphosphatemia and 1,25-Dihydroxyvitamin D in Vascular Calcification and Mortality in Fibroblastic Growth Factor 23 Null Mice. J Am Soc Nephrol, 2007; 18: 2116-2124
- 20) Hesse M, Frohlich LF, Zeitz U, Lanske B and Erben RG: Ablation of vitamin D signaling rescues bone, mineral, and glucose homeostasis in Fgf-23 deficient mice. Matrix Biol, 2007; 26: 75-84
- 21) Razzaque MS, Sitara D, Taguchi T, St-Arnaud R and Lanske B: Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. Faseb J, 2006; 20: 720-722
- 22) Ohnishi M, Nakatani T, Lanske B and Razzaque MS: Reversal of mineral ion homeostasis and soft-tissue calcification of klotho knockout mice by deletion of vitamin D 1alpha-hydroxylase. Kidney Int, 2009; 75: 1166-1172
- 23) Kuro-o M: A potential link between phosphate and aging--lessons from Klotho-deficient mice. Mech Ageing Dev, 2010; 131: 270-275
- 24) Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G, Cholesterol and Recurrent Events Trial I: Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation, 2005; 112: 2627-2633
- 25) Stenvinkel P, Painer J, Kuro-o M, Lanaspa M, Arnold W, Ruf T, Shiels PG and Johnson RJ: Novel treatment strategies for chronic kidney disease: insights from the animal kingdom. Nat Rev Nephrol, 2018; 14: 265-284
- 26) Kuro-o M and Moe OW: FGF23-alphaKlotho as a paradigm for a kidney-bone network. Bone, 2016; 100: 4-18
- 27) Jahnen-Dechent W, Büscher A, Köppert S, Heiss A, Kuro-o M and Smith ER: Mud in the blood the role of protein-mineral complexes and extracellular vesicles in biomineralisation and calcification. J Struct Biol, 2020; 107577
- 28) Heiss A, DuChesne A, Denecke B, Grotzinger J, Yamamoto K, Renne T and Jahnen-Dechent W: Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J Biol Chem, 2003; 278: 13333-13341
- 29) Herrmann M, Schafer C, Heiss A, Graber S, Kinkeldey A, Buscher A, Schmitt MM, Bornemann J, Nimmerjahn F, Helming L, Gordon S and Jahnen-Dechent W: Clearance of Fetuin-A--Containing Calciprotein Particles is Mediated by Scavenger Receptor-A. Circ Res, 2012;
- 30) Akiyama K, Miura Y, Hayashi H, Sakata A, Matsumura Y, Kojima M, Tsuchiya K, Nitta K, Shiizaki K, Kurosu H and Kuro-o M: Calciprotein particles regulate fibroblast growth factor-23 expression in osteoblasts. Kidney Int, 2019; 97: 702-712
- 31) Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J and Hebert SC: Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature, 1993; 366: 575-580
- 32) Quinn SJ, Thomsen AR, Pang JL, Kantham L, Brauner-Osborne H, Pollak M, Goltzman D and Brown EM: Interactions between calcium and phosphorus in the regulation of the production of fibroblast growth factor 23 in vivo. Am J Physiol Endocrinol Metab, 2013; 304: E310-320
- 33) Rodriguez-Ortiz ME, Lopez I, Munoz-Castaneda JR, Martinez-Moreno JM, Ramirez AP, Pineda C, Canalejo A, Jaeger P, Aguilera-Tejero E, Rodriguez M, Felsenfeld A and Almaden Y: Calcium deficiency reduces circulating levels of FGF23. J Am Soc Nephrol, 2012; 23: 1190-1197
- 34) Aghagolzadeh P, Bachtler M, Bijarnia R, Jackson C, Smith ER, Odermatt A, Radpour R and Pasch A: Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-alpha. Atherosclerosis, 2016; 251: 404-414
- 35) Smith ER, Hanssen E, McMahon LP and Holt SG: Fetuin-A-containing calciprotein particles reduce mineral stress in the macrophage. PloS one, 2013; 8: e60904
- 36) Hamano T, Matsui I, Mikami S, Tomida K, Fujii N, Imai E, Rakugi H and Isaka Y: Fetuin-mineral complex reflects extraosseous calcification stress in CKD. J Am Soc Nephrol, 2010; 21: 1998-2007
- 37) Smith ER, Ford ML, Tomlinson LA, Rajkumar C, McMahon LP and Holt SG: Phosphorylated fetuin-A-containing calciprotein particles are associated with aortic stiffness and a procalcific milieu in patients with pre-dialysis CKD. Nephrol Dial Transplant, 2012; 27: 1957-1966
- 38) Miura Y, Iwazu Y, Shiizaki K, Akimoto T, Kotani K, Kurabayashi M, Kurosu H and Kuro-o M: Identification and quantification of plasma calciprotein particles with distinct physical properties in patients with chronic kidney disease. Scientific reports, 2018; 8: 1256
- 39) Franceschi C and Campisi J: Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. The journals of gerontology Series A, Biological sciences and medical sciences, 2014; 69 Suppl 1: S4-9
- 40) Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E and De Benedictis G: Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci, 2000; 908: 244-254
- 41) Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, Saretzki G, Fox C, Lawless C, Anderson R, Hewitt G, Pender SL, Fullard N, Nelson G, Mann J, van de Sluis B, Mann DA and von Zglinicki T: Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nature communications, 2014; 2: 4172
- 42) Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD and Ross OA: Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front Immunol, 2018; 9: 586
- 43) Zhao G, Xu MJ, Zhao MM, Dai XY, Kong W, Wilson GM, Guan Y, Wang CY and Wang X: Activation of nuclear factor-kappa B accelerates vascular calcification by inhibiting ankylosis protein homolog expression. Kidney Int, 2012; 82: 34-44
- 44) Voelkl J, Tuffaha R, Luong TTD, Zickler D, Masyout J, Feger M, Verheyen N, Blaschke F, Kuro-o M, Tomaschitz A, Pilz S, Pasch A, Eckardt KU, Scherberich JE, Lang F, Pieske B and Alesutan I: Zinc Inhibits Phosphate-Induced Vascular Calcification through TNFAIP3-Mediated Suppression of NF-kappaB. J Am Soc Nephrol, 2018; 29: 1636-1648
- 45) Ter Braake AD, Smit AE, Bos C, van Herwaarden AE, Alkema W, van Essen HW, Bravenboer N, Vervloet MG, Hoenderop JGJ and de Baaij JHF: Magnesium prevents vascular calcification in Klotho deficiency. Kidney Int, 2020; 97: 487-501
- 46) Sakaguchi Y, Hamano T, Obi Y, Monden C, Oka T, Yamaguchi S, Matsui I, Hashimoto N, Matsumoto A, Shimada K, Takabatake Y, Takahashi A, Kaimori JY, Moriyama T, Yamamoto R, Horio M, Yamamoto K, Sugimoto K, Rakugi H and Isaka Y: A Randomized Trial of Magnesium Oxide and Oral Carbon Adsorbent for Coronary Artery Calcification in Predialysis CKD. J Am Soc Nephrol, 2019; 30: 1073-1085
- 47) Pasch A, Farese S, Graber S, Wald J, Richtering W, Floege J and Jahnen-Dechent W: Nanoparticle-based test measures overall propensity for calcification in serum. J Am Soc Nephrol, 2012; 23: 1744-1752
- 48) Ewence AE, Bootman M, Roderick HL, Skepper JN, McCarthy G, Epple M, Neumann M, Shanahan CM and Proudfoot D: Calcium phosphate crystals induce cell death in human vascular smooth muscle cells: a potential mechanism in atherosclerotic plaque destabilization. Circ Res, 2008; 103: e28-34
- 49) Scott CC, Vacca F and Gruenberg J: Endosome maturation, transport and functions. Semin Cell Dev Biol, 2014; 31: 2-10
- 50) Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, Furmanik M, Sanchis P, De Rosales RT, Alvarez-Hernandez D, Shroff R, Yin X, Muller K, Skepper JN, Mayr M, Reutelingsperger CP, Chester A, Bertazzo S, Schurgers LJ and Shanahan CM: Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res, 2015; 116: 1312-1323
- 51) Villa-Bellosta R and Sorribas V: Phosphonoformic acid prevents vascular smooth muscle cell calcification by inhibiting calcium-phosphate deposition. Arterioscler Thromb Vasc Biol, 2009; 29: 761-766
- 52) Goldstein JL and Brown MS: A century of cholesterol and coronaries: from plaques to genes to statins. Cell, 2015; 161: 161-172
- 53) Stenvinkel P and Larsson TE: Chronic Kidney Disease: A Clinical Model of Premature Aging. Am J Kidney Dis, 2013; 62: 339-351
- 54) Kenyon CJ: The genetics of ageing. Nature, 2010; 464: 504
- 55) López-Lluch G and Navas P: Calorie restriction as an intervention in ageing. J Physiol, 2016; 594: 2043-2060
- 56) Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL and van Deursen JM: Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature, 2011; 479: 232-236
- 57) Madeo F, Carmona-Gutierrez D, Hofer SJ and Kroemer G: Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab, 2019; 29: 592-610
- 58) Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ and Robbins PD: The Clinical Potential of Senolytic Drugs. J Am Geriatr Soc, 2017; 65: 2297-2301
- 59) Kuro-o M: Phosphate and Klotho. Kidney Int, 2011; 79: S20-23
- 60) Chopra A and Lineweaver CH: The major elemental abundance differences between life, the oceans, and tha Sun. In: Proceedings of the 8th Australian Space Science Conference, ed by Short W and Cairns I, pp49-55, National Space Society of Australia Ltd, Sydney, Australia, 2008



