Abstract
Aim: Genetic testing can provide a definitive diagnosis of familial hypercholesterolemia (FH). However, accessibility of genetic testing may be limited in certain countries where it is not considered “standard of care,” including Japan. In addition, mutations responsible for FH cannot be identified in approximately 30% of patients.
Methods: EXPLORE-J is a multicenter, prospective, observational study of patients presenting with acute coronary syndrome (ACS). The genetic data were analyzed and adjudicated as pathogenic, indeterminate, or nondetectable pathogenic variant.
Results: Of 1,944 patients, 431 underwent genetic screening. Overall, most patients had nonpathogenic variants of LDLR, LDLRAP1, or PCSK9 (n=396, 91.9%). Of the 25 (5.8%) patients with pathogenic variants, variants of the LDLR gene and the PCSK9 gene were seen in 10 and 15 patients, respectively. Indeterminate variants were observed in 10 (2.3%) patients. Of the 431 patients, eight (1.9%) met the criteria for a diagnosis of FH using the Japanese Atherosclerosis Society (JAS) 2017 guidelines. When genetic data were incorporated, 33 (7.7%) patients met the JAS guidelines. No patients with FH pathogenic variants satisfied the JAS clinical criteria for a diagnosis of FH.
Conclusions: The results revealed a higher prevalence of genetic mutations of FH among Japanese patients with ACS and a low sensitivity of the FH diagnostic criteria of the JAS 2017 guidelines. These findings highlight the difficulties of FH diagnosis in patients with ACS in the acute phase and suggest the importance of genetic testing and family history.
Introduction
Familial hypercholesterolemia (FH) is an inherited autosomal dominant disease that is characterized by severely elevated low-density lipoprotein cholesterol (LDL-C), early-onset coronary artery disease (CAD), and tendon or cutaneous xanthomas1-3). The prevalence of FH worldwide is approximately 1 in 200–500, and the estimated number of cases of FH in Japan is 300,000 4). However, in Japan, less than 1% of those with FH are estimated to have been diagnosed using the Dutch Lipid Clinical Network guidelines5).
The diagnosis of FH may include a combination of family history, clinical signs (e.g., tendon xanthomas), and LDL-C levels. Although there are several guidelines for the clinical diagnosis of FH, including the Japan Atherosclerosis Society (JAS) 2017 guidelines6), the Dutch Lipid Clinical Network guidelines5), and the Simon Broome Register guidelines7), there are differences between these guidelines in the criteria used to diagnose FH. The Dutch Lipid Clinical Network and Simon Broome Register guidelines include genetic testing, whereas the JAS 2017 guidelines currently do not.
Genetic testing can provide a definite diagnosis of FH by detection of pathogenic mutations in the genes coding for the low-density lipoprotein receptor (LDLR), apolipoprotein B (apoB), and proprotein convertase subtilisin/kexin type 9 (PCSK9)8-10), or a rare recessive form of FH, autosomal recessive hypercholesterolemia, which is caused by loss-of-function mutations in the LDLR adaptor protein-1 (LDLRAP1)8, 11). However, accessibility of genetic testing may be limited in countries where it is not considered “standard of care,” including Japan12), and in up to 30% of cases a mutation cannot be identified13).
EXPLORE-J is a prospective, large-scale, observational study using data from an acute coronary syndrome (ACS) registry, conducted at 59 centers in Japan14). ACS encompasses a range of conditions compatible with acute myocardial ischemia and/or myocardial infarction (MI), including ST-segment elevation MI, non–ST-segment elevation MI, and unstable angina15). Large observational studies in Europe have investigated the prevalence of FH in patients with ACS, including genetically confirmed cases16, 17). However, the evidence base for Japan is lacking, and the JAS 2017 guidelines have higher cut-off values for LDL-C and Achilles tendon thickness (ATT) when identifying possible FH3, 14). To date, results from the EXPLORE-J study have highlighted the prevalence of FH in patients with ACS in Japan18), as well as described lipid management at the time of ACS19). Using the JAS 2017 guidelines, FH was approximately five times more prevalent in patients with ACS than in the general population18).
Aim
The overall aim of EXPLORE-J was to evaluate lipid management and persistent cardiovascular risk in patients hospitalized for ACS, with a focus on underlying hypercholesterolemia and FH14). The current analysis of the EXPLORE-J registry aimed to identify the characteristics of patients with confirmed pathogenic variants associated with FH. In addition, this analysis aimed to evaluate FH prevalence in patients with ACS, both with and without genetic testing, according to the JAS 2017 guidelines, the Dutch Lipid Clinical Network guidelines, and the Simon Broome Register guidelines.
Methods
Study Design and Patients
EXPLORE-J was a multicenter, prospective, observational study that was conducted between April 2015 and August 2018. Full details of the methods have been published previously14). In brief, patients with ACS who required hospitalization were recruited and registered at 59 sites between April 2015 and August 2016. Patients were then followed up for 2 years. Baseline data, including LDL-C levels, presence of xanthoma, and family history, were reported at Visit 1, within 14 days after hospitalization due to ACS.
Genetic Examinations
Separate informed consent was obtained for genetic examinations. Single nucleotide variations or insertion/deletion in genes encoding key proteins were evaluated. Mutations in genes encoding key proteins involved in the LDLR endocytic and recycling pathways were analyzed (LDLR; LDLRAP1; PCSK9). The APOB gene was not analyzed because no cases of APOB mutations had been previously reported in the Japanese population when the analysis was conducted6). Genetic mutations were categorized as nonpathogenic variant (−), indeterminate pathogenic variant (±, not reported previously, but potentially pathogenic), and pathogenic variant (+, reported pathogenicity of FH). The variants detected in the LDLR or PCSK9 genes were classified as pathogenic using ClinVar, the Leiden Open Variation database, and population data from the Exome Aggregation Consortium, the Japanese Human Genetic Variation Database, and the Tohoku Medical Megabank Organization, as well as in silico tools/software, and functional data based on guidelines edited by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Diagnosis of FH
In this study, three sets of diagnostic guidelines for FH were used for each patient: the JAS 2017 guidelines6), the Dutch Lipid Clinical Network guidelines5), and the Simon Broome guidelines7). Details of each are shown in Table 1. Diagnoses of FH by the JAS 2017 guidelines were conducted both with and without genetic testing as a definitive criterion (identification of a pathogenic variant led to a positive FH diagnosis; functional variants in LDLR and PCSK9 genes were screened in EXPLORE-J).
Table 1.
Diagnostic guidelines for FH
| Diagnostic guidelines |
Criteria |
| The Japanese Atherosclerosis Society guidelines 6)
|
At least two of the following:
|
| ・Hyper LDL-C (≥ 180 mg/dL before treatment) |
| - Secondary hyperlipidemia is excluded |
| - Lipid levels before treatment are considered if patients were on medications |
| - FH is strongly suspected if LDL-C is ≥ 250 mg/dL |
|
・Tendon xanthoma (tendon xanthoma in the back of hand, elbow, knee, etc.; or Achilles tendon
thickening ≥ 9 mm) or tuberous xanthoma (excluding xanthelasma of eyelid)
|
| ・Familial history of FH (blood relatives within the second degree of kinship) or early-onset CAD (male subjects aged <55 years; female subjects aged <65 years) |
| Diagnosis by genetic testing is advised if FH is suspected |
| The Dutch Lipid Clinical Network guidelines for FH a 5)
|
Family history: First-degree relative with known premature coronary and vascular disease (1 point); first- degree relative with known LDL-C level above the 95th percentile b (1 point); first-degree relative with tendon xanthoma and/or corneal arcus b (2 points); children aged <18 years with LDL-C level above the 95th percentile b (2 points)
|
| Clinical history: Patient with premature cerebral or peripheral vascular disease (1 point); or patient with premature CAD (2 points)
|
| Physical examination: Corneal arcus prior to age 45 yearsb (4 points); or tendinous xanthoma (6 points)
|
| Genetic testing: Functional mutation in the LDLR, APOB b, or PCSK9 genes (8 points)
|
|
LDL-C levels: 155–189 mg/dL (1 point); 190–249 mg/dL (3 points); 250–329 mg/dL (5 points); ≥ 330
mg/dL (8 points)
|
| The S imon B r oome guidelinesc 7)
|
Criterion 1: Total cholesterol levels > 290 mg/dL or LDL-C > 190 mg/dL in adults; or total cholesterol levels > 260 mg/dL or LDL-C > 155 mg/dL in childrenb
|
| Criterion 2: Tendon xanthomas in the patient, or tendon xanthomas in a first- or second-degree relative b
|
|
Criterion 3: DNA-based evidence of an LDLR mutation, familial defective apoB-100 b, or a PCSK9
mutation
|
| Criterion 4: Family history of myocardial infarction in a second-degree relative <50 years of age or first- degree relative <60 years of age b
|
|
Criterion 5: Family history of elevated total cholesterol > 290 mg/dL in an adult first- or second-degree
relative; or family history of elevated total cholesterol > 260 mg/dL in a child, brother, or sister aged ≤ 16 years b
|
ApoB, apolipoprotein B; CAD, coronary artery disease; FH, familial hypercholesterolemia; LDL-C, low-density lipoprotein cholesterol; LDLR, low-density lipoprotein receptor; PCSK9, proprotein convertase subtilisin/kexin type 9.
a 3–5 points indicate possible FH, 6–8 points indicate probable FH, and > 8 points indicate definite FH.
b Criteria not collected as part of the EXPLORE-J study.
c Criterion 1+ 2 or 3 indicated definitive FH; criterion 1 +4 or 5 indicated possible FH.
The following criteria were assessed as they are included in all three guidelines for the diagnosis of FH: LDL-C, ATT, and family history of CAD. The highest available measurement of LDL-C obtained at or before Visit 1 was used for the diagnosis of FH. LDL-C was also measured (calculated or direct) prior to hospitalization without treatment (for patients who had available LDL-C values off statin treatment any time before hospitalization) and after hospitalization. In this study, both calculated (e.g., by the Friedewald equation) and/or direct (e.g., by homogeneous direct methods) LDL-C values were collected. For these analyses, calculated LDL-C values were used if available; if calculated LDL-C values were not available, direct LDL-C values were used. ATT was measured at the central reading laboratory by blinded investigators. In addition, family history of CAD was collected at enrollment to help facilitate FH diagnosis.
Statistical Analyses
Baseline characteristics and LDL-C values between genetic variants were described by mean, median, standard deviation, and range for continuous data, and by proportion in each category for categorical data. For comparisons of baseline characteristics and LDL-C values, P-values are provided for baseline characteristics based on Fisher’s exact test for categorical variables and analysis of variance tests for continuous variables. A two-sided P-value of <0.05 was considered significant. Comparisons between diagnostic guidelines for genetic variants are descriptive only. Receiver operating characteristic (ROC) curves were plotted for LDL-C and ATT to FH diagnosis, and cut-off values were obtained using the Youden index20).
Ethics Approval and Consent to Participate
This study was conducted in compliance with the Declaration of Helsinki (amended in October 2013) and the Ethical Guidelines for Medical and Health Research Involving Human Subjects (enacted on December 22, 2014). Prior to the study initiation, the investigator or sub-investigators submitted the protocol and informed consent form to the ethical review committee of each study center and obtained their approval. Patient anonymity was protected by the use of subject identification codes. A co-operation fee of 5000 Japanese yen (approximately US$42 or €37 in October 2015) for study participation was provided for each patient on request from the study center. All patients were required to provide written informed consent.
Results
In total, 1,944 patients were included in the EXPLORE-J study. Of these, 431 underwent genetic testing and were analyzed. Pathogenic variants were observed in 25 (5.8%) patients. Of these 25 patients, 10 (40.0%) had a pathogenic variant of LDLR, and 15 (60.0%) had a pathogenic variant of PCSK9 (Supplementary Table 1). No patients had pathogenic variants of LDLRAP1. Indeterminate variants were observed in 10 (2.3%) patients (Supplementary Table 2). Of these, five (50.0%) patients had indeterminate variants of LDLR and another five (50.0%) had indeterminate variants of PCSK9. Overall, most patients had nonpathogenic variants of LDLR, LDLRAP1, or PCSK9 (n=396, 91.9%).
Supplementary Table 1.
Characteristics of patients with pathogenic variants (
n = 25)
| Age, years |
Sex |
FH
history
|
FH
diagnosis by JAS criteria
|
Premature CAD
family history
|
CAD
family history
|
Highest LDL-C
level, mg/ dL
|
Statin use |
ATT, mm |
Gene |
Mutationa
|
Amino acidb
|
| 50-59 |
Male |
No |
No |
No |
Yes |
119 |
Yes |
7.9 |
PCSK9
|
c.94G> A |
p.E32K |
| 50-59 |
Male |
No |
No |
N/A |
N/A |
136 |
Yes |
6.2 |
LDLR
|
c.344G> A |
p.R115H |
| 60-69 |
Male |
Possible |
No |
N/A |
N/A |
146 |
No |
8.2 |
PCSK9
|
c.94G> A |
p.E32K |
| 50-59 |
Male |
No |
No |
N/A |
N/A |
154 |
No |
6.2 |
LDLR
|
c.344G> A |
p.R115H |
| 70-79 |
Male |
Possible |
No |
No |
No |
78 |
Yes |
8.8 |
PCSK9
|
c.94G> A |
p.E32K |
| 70-79 |
Male |
Possible |
No |
No |
No |
109 |
Yes |
5.8 |
LDLR
|
c.344G> A |
p.R115H |
| 70-79 |
Female |
Possible |
No |
No |
No |
245 |
No |
6.2 |
PCSK9
|
c.94G> A |
p.E32K |
| 60-69 |
Male |
Yes |
No |
No |
Yes |
221 |
No |
5.6 |
PCSK9
|
c.94G> A |
p.E32K |
| 40-49 |
Male |
Yes |
No |
No |
Yes |
168 |
Yes |
8.9 |
LDLR
|
c.1845+2T> Cb
|
N/A |
| 80-89 |
Male |
No |
No |
No |
No |
136 |
No |
6.9 |
PCSK9
|
c.94G> A |
p.E32K |
| 60-69 |
Male |
No |
No |
No |
No |
204 |
No |
6.1 |
PCSK9
|
c.94G> A |
p.E32K |
| 60-69 |
Male |
No |
No |
No |
No |
151 |
No |
N/A |
PCSK9
|
c.94G> A |
p.E32K |
| 70-79 |
Female |
Yes |
No |
Yes |
Yes |
113 |
Yes |
7.8 |
LDLR
|
c.810C> Ac
|
p.C270X |
| 50-59 |
Female |
Yes |
No |
No |
Yes |
290 |
No |
5.3 |
PCSK9
|
c.1486C> T |
p.R496W |
| 60-69 |
Female |
No |
No |
No |
No |
98 |
Yes |
7.4 |
LDLR
|
c.1784G> A |
p.R595Q |
| 70-79 |
Female |
Yes |
No |
No |
No |
153 |
Yes
(plus ezetimibe)
|
17.5 |
LDLR
|
c.2390-2A> Td
|
N/A |
| 60-69 |
Male |
No |
No |
No |
Yes |
126 |
No
(plus fibrate)
|
5.7 |
PCSK9
|
c.94G> A |
p.E32K |
| 60-69 |
Male |
No |
No |
No |
Yes |
188 |
No |
6.4 |
LDLR
|
c.1747C> T |
p.H583Y |
| 30-39 |
Male |
Possible |
No |
No |
Yes |
300 |
Yes |
8.2 |
PCSK9
|
c.94G> A |
p.E32K |
| 60-69 |
Male |
No |
No |
N/A |
N/A |
103 |
No |
6.7 |
LDLR
|
c.1783C> T |
p.R595W |
| 60-69 |
Male |
No |
No |
N/A |
N/A |
173 |
No |
6.3 |
PCSK9
|
c.94G> A |
p.E32K |
| 70-79 |
Male |
No |
No |
N/A |
N/A |
122 |
No |
N/A |
LDLR
|
c.1702C> G |
p.L568V |
| 70-79 |
Male |
No |
No |
N/A |
N/A |
121 |
No |
7.2 |
PCSK9
|
c.94G> A |
p.E32K |
| 60-69 |
Female |
Yes |
No |
No |
No |
108 |
Yes |
12.8 |
PCSK9
|
c.94G> A |
p.E32K |
| 60-69 |
Male |
No |
No |
No |
No |
167 |
No |
6.8 |
PCSK9
|
c.94G> A |
p.E32K |
ATT, Achilles tendon thickness; CAD, coronary artery disease; FH, familial hypercholesterolemia; JAS, Japanese Atherosclerosis Society; LDL-C, low-density lipoprotein cholesterol; N/A, not available.
a Mutations were missense mutations, except for splice donor variant, stop codon mutation, and splice acceptor variant (denoted by b , c , and d ,respectively).
b Amino acid abbreviations: C, cysteine; E, glutamic acid; H, histidine; K, lysine; L, leucine; Q, glutamine; R, arginine; V, valine; W, tryptophan; X, stop codon; Y, tyrosine.
Supplementary Table 2.
Characteristics of patients with indeterminate variants (
n = 10)
| Age |
Sex |
FH
history
|
FH
diagnosis by JAS criteria
|
Premature CAD
family historya
|
CAD
family history
|
Highest LDL-C
level
|
Statin use |
ATT, mm |
Gene |
Mutation |
Amino acidb
|
| 50-59 |
Male |
No |
No |
No |
No |
141 |
No |
7.7 |
PCSK9
|
c.1765G> A |
p.V589M |
| 60-69 |
Male |
Possible |
No |
No |
No |
191 |
No |
8.7 |
LDLR
|
c.211G> A |
p.G71R |
| 60-69 |
Male |
No |
No |
N/A |
N/A |
142 |
No |
6.3 |
LDLR
|
c.211G> A |
p.G71R |
| 70-79 |
Male |
No |
No |
Yes |
Yes |
115 |
No |
N/A |
PCSK9
|
c.212C> T |
p.P71L |
| 80-89 |
Female |
Yes |
No |
No |
No |
112 |
No |
6.6 |
PCSK9
|
c.1954A> G |
p.N652D |
| 80-89 |
Male |
Yes |
No |
No |
No |
91 |
No |
5.3 |
PCSK9
|
c.1975C> G |
p.R659G |
| 80-89 |
Female |
No |
No |
N/A |
N/A |
89 |
No |
N/A |
PCSK9
|
c.1727C> T |
p.P576L |
| 70-79 |
Female |
No |
No |
No |
No |
166 |
No |
6.2 |
LDLR
|
c.2257C> T |
p.P753S |
| 50-59 |
Male |
No |
No |
No |
Yes |
158 |
Yes |
7.4 |
LDLR
|
c.1546G> A |
p.G516S |
| 50-59 |
Male |
Possible |
No |
No |
No |
150 |
No |
10.2 |
LDLR
|
c.1834G> T |
p.A612S |
ATT, Achilles tendon thickness; CAD, coronary artery disease; FH, familial hypercholesterolemia; JAS, Japanese Atherosclerosis Society; LDL-C, low-density lipoprotein cholesterol; N/A, not available.
a Male <55 years, female <65 years.
b Amino acid abbreviations: A, alanine; D, aspartic acid; G glycine; L, leucine; M, methionine; N, asparagine; P, proline; R, arginine; S, serine; V, valine.
Baseline characteristics are shown in Table 2. Overall, the baseline characteristics of the 431 patients who underwent genetic testing were similar to those previously reported for the entire EXPLORE-J population (mean [standard deviation (SD)] age, 66.0 [12.2] years; men, 80.3%; mean [SD] body mass index, 24.2 [3.6] kg/m2)18). Patients with pathogenic variants had generally higher LDL-C levels at baseline compared with patients with nonpathogenic or indeterminate variants before hospitalization (without medication) and after hospitalization (Table 3). Other baseline characteristics were similar between the genetic variants, with no difference observed for any baseline characteristic (Table 2; Table 3).
Table 2.
Baseline characteristics by genetic variants
18)
|
Nonpathogenic variant (–) (n = 396)
|
Indeterminate variant (±) (n = 10)
|
Pathogenic variant (+) (n = 25)
|
Total (n = 431)
|
P-value
|
| Age, years, mean (SD) |
65.7 (11.7) |
69.7 (12.2) |
63.6 (10.8) |
65.7 (11.7) |
0.378 |
| Male, n (%)
|
317 (80.1) |
7 (70.0) |
19 (76.0) |
343 (79.6) |
0.528 |
| Female, n (%)
|
79 (19.9) |
3 (30.0) |
6 (24.0) |
88 (20.4) |
|
| BMI, kg/m2, mean (SD)
|
24.3 (3.8) a
|
24.1 (3.3) |
23.0 (3.3) |
24.3 (3.8) b
|
0.221 |
| Weight, kg, mean (SD) |
65.5 (13.4) a
|
64.0 (15.9) |
62.9 (12.7) |
65.3 (13.4) b
|
0.633 |
| STEMI, n (%)
|
270 (68.2) |
7 (70.0) |
18 (72.0) |
295 (68.4) |
0.732 |
| NSTEMI, n (%)
|
52 (13.1) |
1 (10.0) |
1 (4.0) |
54 (12.5) |
|
| Unstable angina, n (%)
|
74 (18.7) |
2 (20.0) |
6 (24.0) |
82 (19.0) |
|
| Diabetes mellitus, n (%)
|
144 (36.4) |
3 (30.0) |
6 (24.0) |
153 (35.5) |
0.446 |
| Hypertension, n (%)
|
305 (77.0) |
10 (100.0) |
17 (68.0) |
332 (77.0) |
0.123 |
| Dyslipidemia, n (%)
|
306 (77.3) |
9 (90.0) |
23 (92.0) |
338 (78.4) |
0.152 |
| Therapy before hospitalization, n (%)
|
|
|
|
|
|
| Statin |
97 (24.5) |
1 (10.0) |
10 (40.0) |
108 (25.1) |
0.133 |
| Intensive statinc
|
16 (4.0) |
0 (0.0) |
0 (0.0) |
16 (3.7) |
0.449 |
| Ezetimibe |
4 (1.0) |
0 (0.0) |
1 (4.0) |
5 (1.2) |
0.347 |
| Statin + ezetimibe |
0 (0.0) |
0 (0.0) |
1 (4.0) |
1 (0.2) |
- |
BMI, body mass index; NSTEMI, non–ST-elevation myocardial infarction; SD, standard deviation; STEMI, ST-elevation myocardial infarction. Baseline characteristics were collected at Visit 1.
a n = 395. b n = 430. c Atorvastatin ≥ 20 mg, rosuvastatin ≥ 10 mg, and pitavastatin ≥ 4 mg.
Table 3.
LDL-C levels by genetic variants
| LDL-C levels, mg/dL, mean (SD) |
Nonpathogenic variant (–) (n = 396)
|
Indeterminate variant (±) (n = 10)
|
Pathogenic variant (+) (n = 25)
|
Total (n = 431)
|
P-value
|
| n
|
392 |
10 |
25 |
427 |
|
| Maximum value at baseline |
125.1 (41.0) |
135.5 (33.2) |
157.2 (57.3) |
127.2 (42.5) |
<0.001 |
| n
|
88 |
5 |
3 |
96 |
|
| Measurement without medication before hospitalization |
131.5 (37.4) |
137.4 (41.3) |
179.7 (99.2) |
133.3 (40.4) |
0.123 |
| n
|
374 |
10 |
24 |
408 |
|
| First measurement after hospitalization |
119.4 (40.4) |
126.9 (38.0) |
148.1 (51.9) |
121.2 (41.6) |
0.004 |
| n
|
346 |
10 |
24 |
380 |
|
| Direct method |
93.3 (31.1) |
107.7 (27.7) |
130.9 (51.1) |
96.1 (33.8) |
<0.001 |
| n
|
343 |
10 |
24 |
377 |
|
| Calculated |
91.9 (31.3) |
106.6 (27.4) |
124.3 (38.9) |
94.4 (32.7) |
<0.001 |
LDL-C, low-density lipoprotein cholesterol; SD, standard deviation.
The percentage of patients meeting each of the three sections (family history of early-onset CAD, ATT, maximum baseline LDL-C levels) of the JAS guidelines by genetic variant is shown in Table 4. Family history was available for most patients across all genetic variants (overall, 77 [17.9%] patients had unknown [information unavailable] family history of early-onset CAD).
Table 4.
The percentage of patients meeting the JAS guideline criteria by genetic variants
| JAS guideline criteria |
|
All patients (n = 431)
|
Nonpathogenic variants (–) and indeterminate variants (±) (n = 406)
|
Pathogenic variants (+) (n = 25)
|
| Family history of early-onset CADa
|
No |
325 (75.4) |
308 (75.9) |
17 (68.0) |
|
Yes |
29 (6.7) |
28 (6.9) |
1 (4.0) |
|
Unknown |
77 (17.9) |
70 (17.2) |
7 (28.0) |
| ATT |
<9 mm |
358 (83.1) |
337 (83.0) |
21 (84.0) |
|
≥ 9 mm |
19 (4.4) |
17 (4.2) |
2 (8.0) |
|
Unknown |
54 (12.5) |
52 (12.8) |
2 (8.0) |
| Baseline LDL-C levels (maximum value) |
<180 mg/dL |
384 (89.1) |
385 (89.9) |
19 (76.0) |
|
≥ 180 mg/dL |
43 (10.0) |
37 (9.1) |
6 (24.0) |
|
Unknown |
4 (0.9) |
4 (1.0) |
0 (0.0) |
a Male <55 years, female <65 years.
ATT, Achilles tendon thickness; CAD, coronary artery disease; JAS, Japan Atherosclerosis Society; LDL-C, low-density lipoprotein cholesterol.
ATT was available for most patients across all genetic variants (overall, 54 [12.5%] patients had unknown ATT [ATT unavailable]). Baseline LDL-C was available for most patients (overall, four [0.9%] patients had unknown baseline LDL-C [baseline LDL-C unavailable]).
Of the patients with known pathogenic variants (n=25; Supplementary Table 1), none of the patients would have been diagnosed with FH according to the JAS 2017 guidelines. Similarly, of the patients with indeterminate variants (n=10; Supplementary Table 2), no patients would have been diagnosed with FH according to the JAS 2017 guidelines. In addition, two (8.0%) patients with pathogenic variants had ATT ≥ 9 mm; however, their highest LDL-C values were 153 and 108 mg/dL, respectively, which did not meet the LDL-C criterion for FH as per the JAS guidelines.
The percentage of patients meeting criteria for each of the three guidelines for the diagnosis of FH (the JAS guidelines, the Dutch Lipid Clinical Network guidelines, or the Simon Broome guidelines) is shown in Supplementary Fig.1. In total, 398 (92.3%) patients did not have FH according to any of the three guidelines. Thirty-three (7.7%) patients had definitive FH according to at least one of the guidelines, and 17 (3.9%) patients had FH according to both the Dutch Lipid Clinical Network and Simon Broome guidelines. Overall, six (1.4%) patients had definitive FH according to all three guidelines.
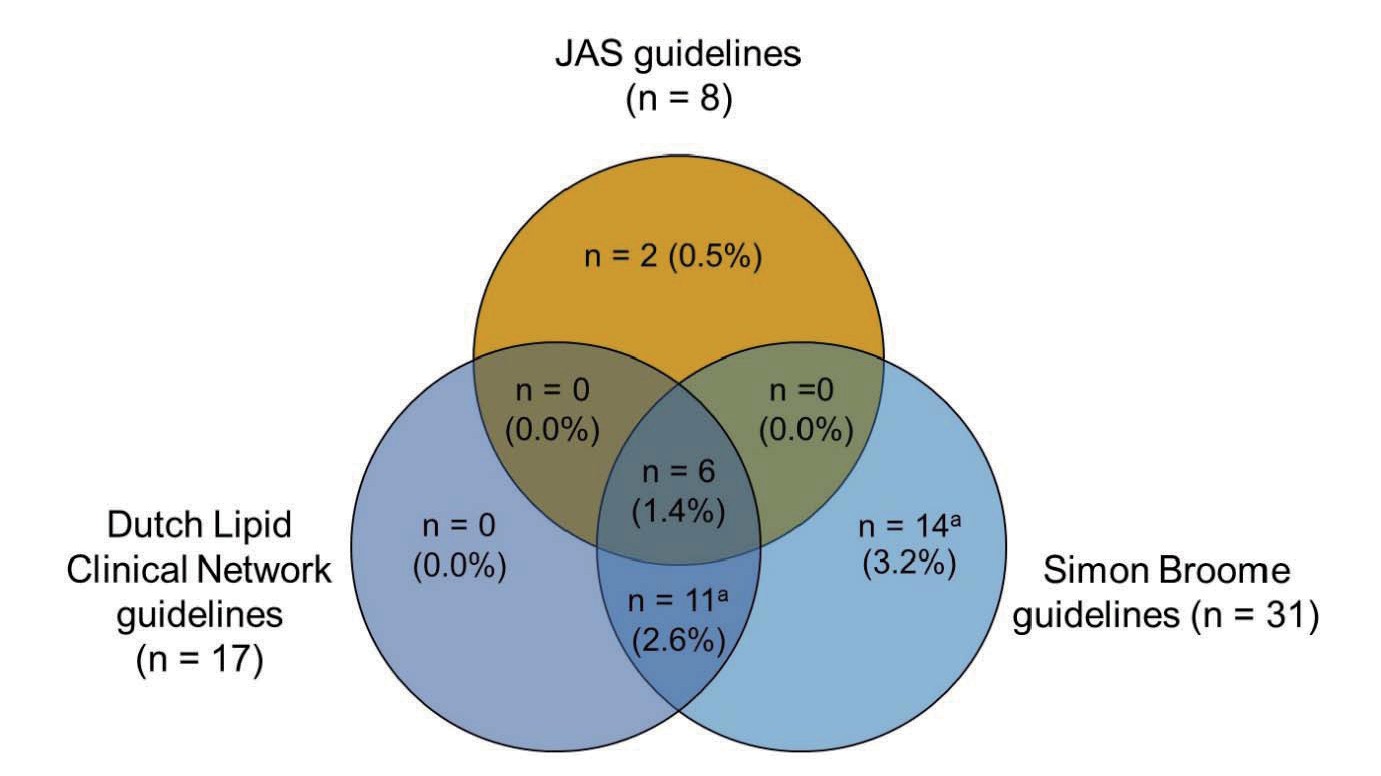
Without genetic testing, only eight (1.9%) of the 431 patients met the JAS 2017 guidelines for FH, compared with definite FH according to the Dutch Lipid Clinical Network guidelines (n=6, 1.4%) or the Simon Broome guidelines (n=6, 1.4%). When the JAS 2017 guidelines were supplemented with genetic data (data not shown), the proportion of patients considered to have FH (n=33, 7.7%) was similar to that of definite FH diagnosed using Dutch Lipid Clinical Network guidelines (n=17, 3.9%) and the Simon Broome guidelines (n=31, 7.2%).
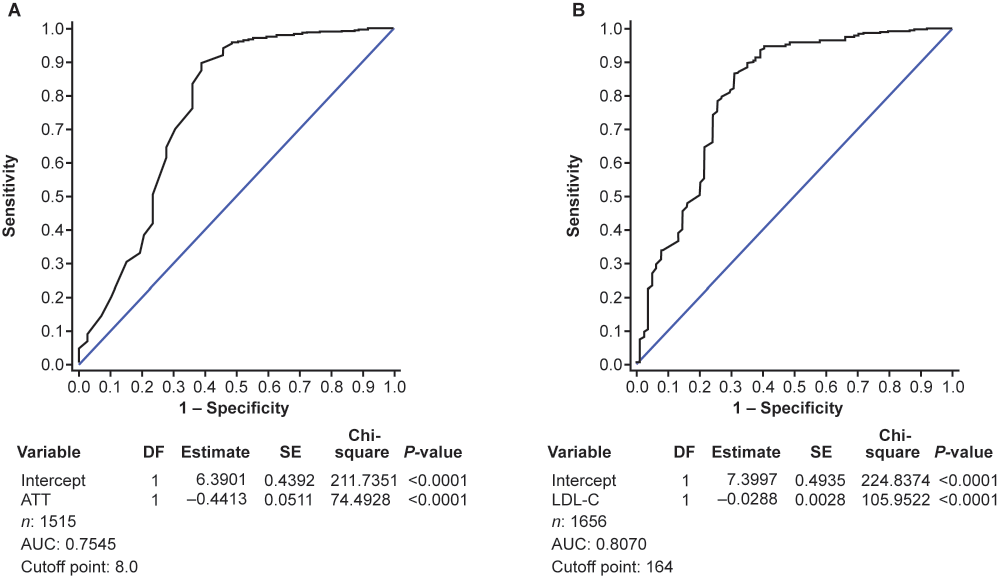
ROC curves of parameters of FH diagnosis via the JAS 2017 guidelines and FH genetic mutations are shown in Fig.1. Cut-offs of LDL-C 164 mg/dL and ATT 8.0 mm have the highest sensitivity and specificity to identify patients with FH when used as part of the JAS guidelines.
Discussion
This analysis of a subgroup of patients with ACS from the EXPLORE-J study who underwent FH genetic testing (n=431) provides several major findings: i) overall, 25 (5.8%) patients had pathogenic variants for FH, mostly in PCSK9; ii) no patients with pathogenic variants met the threshold for FH according to the JAS guidelines; iii) when the JAS guidelines were supplemented with genetic data, the proportion of patients considered to have FH was similar to that of the Dutch Lipid Clinical Network and Simon Broome guidelines; and iv) when using the JAS guidelines for FH, according to the ROC parameter of values analysis, cut-off values of LDL-C 164 mg/dL and ATT 8.0 mm have the highest sensitivity and specificity to identify patients with FH.
Of the 25 patients who had pathogenic variants for FH, many (n=15, 60%) had a pathogenic variant of PCSK9. There were more mutations in the PCSK9 gene (associated with higher risk of CAD) than previously reported in Japanese patients with pathogenic variants associated with FH10, 21). This may be due to differences in the population, region, and family history of the patient samples in the previous studies. Additionally, the prevalence of the E32K variant of PCSK9 in Japanese patients with pathogenic variants for FH may be higher than that in other countries22-24). Patients who have this mutation show milder phenotypes compared with those with LDLR mutations25). Therefore, it may be possible that some Japanese FH patients with this mutation do not meet the criteria of FH in the JAS 2017 guidelines for this reason and are therefore not diagnosed as having FH.
It is surprising that none of the patients with pathogenic variants would be diagnosed with FH using the JAS 2017 guidelines. Their LDL-C levels were much lower than those of statin-naïve FH patients without ACS. One reason for the lack of patients with pathogenic variants meeting the JAS guidelines’ definition of FH could be that 40% of these patients were receiving a statin. Another reason is that LDL-C levels decrease during the acute phase of ACS. It has been reported that LDL-C can decrease by up to 48% after acute MI26). These may have masked their untreated LDL-C levels by lowering LDL-C below the 180 mg/dL threshold required to identify FH with the JAS 2017 guidelines. Cut-off levels of LDL-C for the diagnosis of FH in the JAS guidelines were determined by using the pre-treatment LDL-C levels in patients with an FH diagnosis and those without27). Furthermore, the JAS guidelines criteria are not designed for patients in the acute phase of ACS.
Notably, four patients with pathogenic variants were reported by their attending physician to have been diagnosed with FH despite not meeting the JAS guidelines for FH during the EXPLORE-J study using ATT. Of these patients, three had LDL-C <180 mg/dL and had statin use prior to the index ACS. This further supports the view that signs of FH might be masked in some patients through prior statin use, especially in this population for whom early initiation of high-intensity statin is recommended or who are already on statins at presentation of ACS6).
Statin use can also mitigate the development of xanthoma and reduce ATT28), and some patients may therefore not have had ATT ≥ 9 mm due to prior statin use. A previous study in Japanese patients with ACS (n=296) demonstrated a prevalence of statin use similar to that in the present study (23.3% vs. 25.1%, respectively)29). However, the detection rate of ATT ≥ 9 mm here (19/377; 5.0%) was lower than that reported by Ohmura et al. (53/296; 17.9%)29). Because not all patients with FH have ATT ≥ 9 mm, this may account for differences between these two studies. Patients in the Ohmura et al. study were from residential areas of Tokyo. In this study, patients were enrolled from all over Japan, which may account for differences29).
In addition, the percentage of patients with FH according to the JAS guidelines reported by Ohmura et al. was 5.7%29), whereas in this study 1.9% of patients would be diagnosed with FH using the JAS 2017 guidelines (without additional genetic testing) and 7.7% would be diagnosed with FH when the JAS guidelines were supplemented with genetic testing29). FH status was determined using the JAS 2017 guidelines with and without additional genetic testing. When genetic testing was incorporated with the JAS 2017 guidelines, the proportion of patients identified as having FH was similar to those diagnosed with definite FH by the Dutch Lipid Clinical Network and Simon Broome guidelines (3.9% and 7.2%, respectively). This suggests that the JAS guidelines without genetic testing might be insufficient for diagnosis of FH in patients with ACS in Japan. In addition, the cut-off point of LDL-C and ATT was analyzed using ROC curves of parameters. According to these ROC curves, we propose that cut-off values of LDL-C 165 mg/dL and ATT 8.0 mm could lead to more accurate diagnosis of FH if incorporated into the current JAS guidelines.
Because of variability in LDL-C after ACS, availability of genetic testing in patients with suspected FH is important in this population. Currently, there are a limited number of hospitals in Japan that can conduct genetic testing for FH, and genetic testing for FH is not reimbursed by health insurance providers6). Stigma of genetic disease (e.g., social perceptions and fears of insurance denial) and lack of resources (such as accessibility to geneticists and genetic counseling) may also hinder diagnosis of FH30, 31). These barriers need to be removed to improve the diagnostic rate of FH and allow cascade screening, which will lead to appropriate lipid-lowering therapy with high-dose statins, ezetimibe, and PCSK9 inhibitors for these patients, lowering their risk of premature cardiovascular events. However, although genetic testing will improve diagnosis in this population, many patients with FH will not have an identifiable mutation. Therefore, clinical evaluation of those presenting with ACS remains crucial.
Interestingly, although a direct comparison may not be meaningful because of the small sample size, a lower proportion of patients with pathogenic variants had a family history of CAD than had those without pathogenic variants (4.0% vs. 6.9%). This may be due to underreporting or the fact that patients with pathogenic variants (and their families) have severe enough hypercholesterolemia that they are already treated with statins, thereby reducing CAD.
This study is limited by the nature of the observational study design. In this analysis, untreated LDL-C levels were only available for three of the patients with pathogenic variants. Ohmura et al. also highlighted the difficulties of collecting LDL-C values for use in the diagnosis of FH and that collection of blood samples varied among the facilities included in their study29). Furthermore, information on family history was unknown for 77 (17.9%) patients overall and seven (28.0%) patients with pathogenic variants. In addition, not all patients received genetic testing, and available data for each criterion in the JAS, Dutch Lipid Clinical Network, and Simon Broome guidelines were not complete for all patients. Furthermore, although APOB mutations are generally not found in the Japanese population, and so were not included in the genetic testing used here, sporadic mutations may occur. In fact, subsequent to this analysis, the first case of FH due to a known pathogenic APOB gene variant was reported in a Japanese family32). Although this study has only examined variants of the LDLR and PCSK9 genes, it might be more informative for FH diagnosis if polygenic variants are examined, such as APOE4 and the sporadic mutations of APOB. Finally, the generalizability of these findings is limited to the Japanese population.
Conclusion
These results revealed genetic mutations of FH found among Japanese patients with ACS. However, to increase the diagnostic accuracy of patients presenting with ACS, the diagnostic thresholds used in the JAS guidelines may need to be re-evaluated for these patients. These findings highlight the importance of genetic testing and family history in the diagnosis of FH in patients with ACS, as well as screening and diagnosis of FH before the initiation of statins in patients that may have FH. A substantial number of high-risk patients could benefit from genetic testing. In addition, we propose that cut-off values of LDL-C 165 mg/dL and ATT 8.0 mm could improve diagnosis of FH if incorporated into the current JAS guidelines.
Acknowledgements
The authors thank the patients, their families, and all investigators involved in the study: Tamio Teramoto of Teikyo University (Tokyo, Japan), Shun Ishibashi of Jichi Medical University (Tochigi, Japan), Kotaro Yokote of Chiba University (Chiba, Japan), Tomonori Okamura of Keio University (Tokyo, Japan), and Hiroyuki Daida of Juntendo University (Tokyo, Japan) for collaboration and advice regarding planning of the study; Mebix (Tokyo, Japan) for assistance with study implementation/operation; BML (Tokyo, Japan) for PCSK9-related and genome-related analysis; CTD (Tokyo, Japan) for consulting; and Shizuya Yamashita of Rinku General Medical Center (Osaka, Japan) and Toru Yoshizumi of Kawasaki Hospital (Hyogo, Japan). The authors would also like to thank Yoshiharu Takagi and Yukiko Morimoto of Sanofi for providing support with statistical analysis; and Yasunori Nakahigashi and Makiko Usami of Sanofi for assistance with study implementation/operation. Medical writing assistance and editorial support, under the direction of the authors, were provided by Kate Carolan, PhD, of Prime Global (Knutsford, United Kingdom) according to Good Publication Practice guidelines (Link) and funded by Sanofi. The authors were involved in the study design as well as the collection, analysis, and interpretation of data. All authors had full access to all the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Financial Support
This study was funded by Sanofi and Regeneron Pharmaceuticals, Inc.
Conflicts of Interest
Mariko Harada-Shiba has received honoraria from Amgen, Astellas, and Sanofi; and research grants from Aegerion, Kaneka and Recordati and held stock of Liid Pharma. Junya Ako has received honoraria from Amgen and Sanofi. Atsushi Hirayama has received honoraria and research grants from Sanofi. Masato Nakamura has received honoraria from Astellas, Amgen, and Sanofi; and research grants from Sanofi. Atsushi Nohara has received honoraria from Sanofi. Kayoko Sato has received research grants from Astellas, Takeda, and Aegerion. Yoshitaka Murakami does not have any conflicts of interest to declare. Ryusuke Koshida was an employee of Sanofi K.K at the time of this study. Asuka Ozaki is an employee of Sanofi K.K. Hidenori Arai has received lecture fees from Daiichi Sankyo, Kowa, MSD, Pfizer, and Sanofi.
References
- 1) Bouhairie VE and Goldberg AC: Familial hypercholesterolemia. Cardiology clinics, 2015; 33: 169-179
- 2) Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD and Wierzbicki AS: Familial hypercholesterolaemia. Nat Rev Dis Primers, 2017; 3: 17093
- 3) Harada T, Inagaki-Tanimura K, Nagao M, Sato Y, Sudo M, Okajima F, Sugihara H and Oikawa S: Frequency of Achilles Tendon Xanthoma in Patients with Acute Coronary Syndrome. J Atheroscler Thromb, 2017; 24: 949-953
- 4) Teramoto T, Sasaki J, Ishibashi S, Birou S, Daida H, Dohi S, Egusa G, Hiro T, Hirobe K, Iida M, Kihara S, Kinoshita M, Maruyama C, Ohta T, Okamura T, Yamashita S, Yokode M, Yokote K, Harada-Shiba M, Arai H, Bujo H, Nohara A, Ohta T, Oikawa S, Okada T and Wakatsuki A: Familial hypercholesterolemia. J Atheroscler Thromb, 2014; 21: 6-10
- 5) Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Boren J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjaerg-Hansen A and European Atherosclerosis Society Consensus Panel: Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J, 2013; 34: 3478-3490a
- 6) Harada-Shiba M, Arai H, Ishigaki Y, Ishibashi S, Okamura T, Ogura M, Dobashi K, Nohara A, Bujo H, Miyauchi K, Yamashita S and Yokote K: Guidelines for Diagnosis and Treatment of Familial Hypercholesterolemia 2017. J Atheroscler Thromb, 2018; 25: 751-770
- 7) Simon Broome Register Group: Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ, 1991; 303: 893-896
- 8) Hovingh GK, Davidson MH, Kastelein JJ and O’Connor AM: Diagnosis and treatment of familial hypercholesterolaemia. Eur Heart J, 2013; 34: 962-971
- 9) Hartgers ML, Ray KK and Hovingh GK: New approaches in detection and treatment of familial hypercholesterolemia. Curr Cardiol Rep, 2015; 17: 109
- 10) Hori M, Ohta N, Takahashi A, Masuda H, Isoda R, Yamamoto S, Son C, Ogura M, Hosoda K, Miyamoto Y and Harada-Shiba M: Impact of LDLR and PCSK9 pathogenic variants in Japanese heterozygous familial hypercholesterolemia patients. Atherosclerosis, 2019; 289: 101-108
- 11) Harada-Shiba M, Takagi A, Miyamoto Y, Tsushima M, Ikeda Y, Yokoyama S and Yamamoto A: Clinical features and genetic analysis of autosomal recessive hypercholesterolemia. J Clin Endocrinol Metab, 2003; 88: 2541-2547
- 12) Nordestgaard BG and Benn M: Genetic testing for familial hypercholesterolaemia is essential in individuals with high LDL cholesterol: who does it in the world? Eur Heart J, 2017; 38: 1580-1583
- 13) Watts GF, Gidding S, Wierzbicki AS, Toth PP, Alonso R, Brown WV, Bruckert E, Defesche J, Lin KK, Livingston M, Mata P, Parhofer KG, Raal FJ, Santos RD, Sijbrands EJ, Simpson WG, Sullivan DR, Susekov AV, Tomlinson B, Wiegman A, Yamashita S, Kastelein JJ and International Familial Hypercholesterolemia Foundation: Integrated guidance on the care of familial hypercholesterolaemia from the International FH Foundation. Eur J Prev Cardiol, 2015; 22: 849-854
- 14) Nakamura M, Uno K, Hirayama A, Ako J, Nohara A, Arai H and Harada-Shiba M: Exploration into lipid management and persistent risk in patients hospitalised for acute coronary syndrome in Japan (EXPLORE-J): protocol for a prospective observational study. BMJ Open, 2017; 7: e014427
- 15) Amsterdam EA, Wenger NK, Brindis RG, Casey DE, Jr., Ganiats TG, Holmes DR, Jr., Jaffe AS, Jneid H, Kelly RF, Kontos MC, Levine GN, Liebson PR, Mukherjee D, Peterson ED, Sabatine MS, Smalling RW and Zieman SJ: 2014 AHA/ACC Guideline for the Management of Patients with Non-ST-Elevation Acute Coronary Syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol, 2014; 64: e139-e228
- 16) Nanchen D, Gencer B, Auer R, Räber L, Stefanini GG, Klingenberg R, Schmied CM, Cornuz J, Muller O, Vogt P, Jüni P, Matter CM, Windecker S, Lüscher TF, Mach F and Rodondi N: Prevalence and management of familial hypercholesterolaemia in patients with acute coronary syndromes. Eur Heart J, 2015; 36: 2438-2445
- 17) Amor-Salamanca A, Castillo S, Gonzalez-Vioque E, Dominguez F, Quintana L, Lluís-Ganella C, Escudier JM, Ortega J, Lara-Pezzi E, Alonso-Pulpon L and Garcia-Pavia P: Genetically Confirmed Familial Hypercholesterolemia in Patients With Acute Coronary Syndrome. J Am Coll Cardiol, 2017; 70: 1732-1740
- 18) Harada-Shiba M, Ako J, Arai H, Hirayama A, Murakami Y, Nohara A, Ozaki A, Uno K and Nakamura M: Prevalence of familial hypercholesterolemia in patients with acute coronary syndrome in Japan: Results of the EXPLORE-J study. Atherosclerosis, 2018; 277: 362-368
- 19) Nakamura M, Ako J, Arai H, Hirayama A, Murakami Y, Nohara A, Uno K, Ozaki A and Harada-Shiba M: Investigation into lipid management in acute coronary syndrome patients from the EXPLORE-J study. J Atheroscler Thromb, 2019; 26: 559-572
- 20) Youden WJ: Index for rating diagnostic tests. Cancer, 1950; 3: 32-35
- 21) Yu W, Nohara A, Higashikata T, Lu H, Inazu A and Mabuchi H: Molecular genetic analysis of familial hypercholesterolemia: spectrum and regional difference of LDL receptor gene mutations in Japanese population. Atherosclerosis, 2002; 165: 335-342
- 22) Noguchi T, Katsuda S, Kawashiri MA, Tada H, Nohara A, Inazu A, Yamagishi M, Kobayashi J and Mabuchi H: The E32K variant of PCSK9 exacerbates the phenotype of familial hypercholesterolaemia by increasing PCSK9 function and concentration in the circulation. Atherosclerosis, 2010; 210: 166-172
- 23) Han SM, Hwang B, Park T-G, Kim D-I, Rhee M-Y, Lee B-K, Ahn YK, Cho BR, Woo J and Hur S-H: Genetic testing of Korean familial hypercholesterolemia using whole-exome sequencing. PloS One, 2015; 10: e0126706
- 24) Marduel M, Carrié A, Sassolas A, Devillers M, Carreau V, Di Filippo M, Erlich D, Abifadel M, Marques‐Pinheiro A and Munnich A: Molecular spectrum of autosomal dominant hypercholesterolemia in France. Human Mutation, 2010; 31: E1811-E1824
- 25) Mabuchi H, Nohara A, Noguchi T, Kobayashi J, Kawashiri MA, Inoue T, Mori M, Tada H, Nakanishi C, Yagi K, Yamagishi M, Ueda K, Takegoshi T, Miyamoto S, Inazu A and Koizumi J: Genotypic and phenotypic features in homozygous familial hypercholesterolemia caused by proprotein convertase subtilisin/kexin type 9 (PCSK9) gain-of-function mutation. Atherosclerosis, 2014; 236: 54-61
- 26) Rosenson RS: Myocardial injury: the acute phase response and lipoprotein metabolism. Journal of the American College of Cardiology, 1993; 22: 933-940
- 27) Harada-Shiba M, Arai H, Okamura T, Yokote K, Oikawa S, Nohara A, Okada T, Ohta T, Bujo H, Watanabe M, Wakatsuki A and Yamashita S: Multicenter study to determine the diagnosis criteria of heterozygous familial hypercholesterolemia in Japan. J Atheroscler Thromb, 2012; 19: 1019-1026
- 28) Tsouli SG, Xydis V, Argyropoulou MI, Tselepis AD, Elisaf M and Kiortsis DN: Regression of Achilles tendon thickness after statin treatment in patients with familial hypercholesterolemia: an ultrasonographic study. Atherosclerosis, 2009; 205: 151-155
- 29) Ohmura H, Fukushima Y, Mizuno A, Niwa K, Kobayashi Y, Ebina T, Kimura K, Ishibashi S and Daida H: Estimated Prevalence of Heterozygous Familial Hypercholesterolemia in Patients With Acute Coronary Syndrome. Int Heart J, 2017; 58: 88-94
- 30) New York-Mid-Atlantic Consortium for Genetic Newborn Screening Services: Understanding genetics: a New York, mid-Atlantic guide for patients and health professionals, Lulu.com, 2009
- 31) Delikurt T, Williamson GR, Anastasiadou V and Skirton H: A systematic review of factors that act as barriers to patient referral to genetic services. Eur J Hum Genet, 2015; 23: 739-745
- 32) Hori M, Takahashi A, Son C, Ogura M and Harada-Shiba M: The first Japanese cases of familial hypercholesterolemia due to a known pathogenic APOB gene variant, c.10580 G>A: p.(Arg3527Gln). Journal of Clinical Lipidology, 2020; 14: 482-486