Abstract
Transitional medicine refers to the seamless continuity of medical care for patients with childhood-onset diseases as they grow into adulthood. The transition of care must be seamless in medical treatment as the patients grow and in other medical aids such as subsidies for medical expenses in the health care system. Inappropriate transitional care, either medical or social, directly causes poorer prognosis for many early-onset diseases, including primary dyslipidemia caused by genetic abnormalities. Many primary dyslipidemias are designated as intractable diseases in the Japanese health care system for specific medical aids, as having no curative treatment and requiring enormous treatment costs for lipid management and prevention of complications. However, there are problems in transitional medicine for primary dyslipidemia in Japan. As for the medical treatment system, the diagnosis rate remains low due to the shortage of specialists, their insufficient link with generalists and other field specialists, and poor linkage between pediatricians and physicians for adults. In the medical care system, there is a mismatch of diagnostic criteria of primary dyslipidemias between children and adults for medical care expense subsidization, as between The Program for the Specific Pediatric Chronic Diseases and the Program for Designated Adult Intractable Diseases. This could lead some patients subsidized in their childhood to no longer be under the coverage of the aids after transition. This review intends to describe these issues in transitional medicine of primary dyslipidemia in Japan as a part of the efforts to resolve the problems by the Committee on Primary Dyslipidemia under the Research Program on Rare and Intractable Disease of the Ministry of Health, Labour and Welfare of Japan.
I Introduction: Why should We Discuss Transitional Medicine for Primary Dyslipidemia?
“Transitional medicine” indicates appropriate treatment processes for childhood-onset illnesses tailored to the patient’s growth and development. Inadequate transitional medicine, especially in congenital metabolic diseases caused by genetic abnormalities, can lead to a poor prognosis for patients. In addition, the connection from pediatric care to adult care must be seamless in the healthcare system including patient aid such as subsidies for medical expenses of chronic intractable diseases. This review article attempts to describe the issues of transitional medicine in primary dyslipidemia in Japan from these two perspectives.
Fig.1 shows the history of the patient aid system for intractable primary dyslipidemias in Japan. In addition, Table 1 summarizes the differences between the pediatric chronic specified disease system and the adult intractable disease system. The Research Program for Treatment of Specific Pediatric Chronic Diseases, which intended to cover medical expenses for congenital metabolic disorders in children, was initiated by the Ministry of Health and Welfare in 1968. The target diseases included “primary hyperlipidemia,” of which the definition was ambiguous because of the immature research stage and understanding of lipoprotein metabolism at that time. As the research progressed in understanding the clinical features and metabolic mechanism of familial hypercholesterolemia (FH) and other diseases, it started to be used for the treatment of pediatric FH. In particular, lipoprotein apheresis therapy, developed at the National Cerebral and Cardiovascular Center (Osaka, Japan) in the early 1980s, was introduced for pediatric homozygous FH. However, as this program was designed only for the youths under the age of 18 (later extended to 20), the highly expensive cost of this chronic life-supporting treatment placed a heavy burden on FH homozygotes as they reached adulthood, forcing a substantial number of the patients to discontinue the treatment. Pushed by this situation, the patient organization of FH was organized to make strong requests to the Ministry of Health, Labor and Welfare to subsidize the medical expense of apheresis treatment of adult FH, supported by the researchers in the National Cerebral and Cardiovascular Center. As a result of these efforts over several years, FH homozygotes were designated in the list for intractable diseases when the Act on Research and Medical Aid on Intractable Diseases was revised in 2009. Thus, transitional medicine became active for FH homozygotes in the Japanese healthcare system.

Table 1.Differences between the Specific Pediatric Chronic Diseases system and the Adult Intractable Diseases system
|
Policy of the Specific Pediatric Chronic Diseases |
Policy of the Adult Intractable Diseases |
| Laws on which the policy is based |
Child Welfare Act |
Act on Medical Care for Patients with Intractable Diseases |
| Requirements for Covered Diseases |
-
1. The disease must be chronic in the course.
-
2. Must be a long-term, life-threatening disease
-
3. A disease whose symptoms and treatment impair quality of life over a long period.
-
4. A disease that continues to cause high medical costs over a long period.
|
-
1. The mechanism of pathogenesis is unclear.
-
2. Lack of established treatment methods.
-
3. Must be a rare disease.
-
4. Requiring long-term medical treatment.
-
5. The number of patients does not reach a certain number (approximately 0.1% of the population or less) in Japan.
-
6. Objective diagnostic criteria must be established.
|
| Number of diseases covered |
788 (As of November 2023) |
338 (As of November 2023) |
| Ages Covered |
Under 18 years of age (however, if continued treatment is deemed necessary, up to under 20 years of age) |
No age limitation |
| Paying one’s expenses |
Subsidies for co-payment of medical insurance (maximum amount to be paid depending on income status) |
Subsidies for co-payment of medical insurance (maximum amount to be paid depending on income status) |
| Implementing Entities |
Prefectures, designated cities, core cities, cities with child guidance centers |
Prefectures, designated cities |
| Related information provided by |
Information Center for Specific Pediatric Chronic Diseases, Japan |
Japan Intractable Diseases Information Center |
| URL |
https://www.shouman.jp/
|
https://www.nanbyou.or.jp/
|
Subsequently, in 2015, the medical expense subsidy system for intractable diseases underwent significant revision, and the list of diseases eligible for medical expense subsidy was overhauled and substantially expanded in both pediatric and adult patients. However, the revision of the list was conducted independently between pediatric and adults, and the development of diagnostic criteria was completed in an extremely short period, without a consensus-building process based on scientific discussions at related academic societies. On the adult side, six new inherited dyslipidemias were added as designated Intractable Diseases eligible to the subsidy system in addition to FH. In pediatrics, previously categorized as “primary hyperlipidemia,” it was divided into six categories less specific except for FH. The newly named diseases for the adult intractable diseases list are lecithin cholesterol acyltransferase (LCAT) deficiency, sitosterolemia, Tangier disease, primary hyperchylomicronemia, cerebrotendinous xanthomatosis, and abetalipoproteinemia. In children, five new categories for Specific Pediatric Chronic Diseases were: primary hyperchylomicronemia, FH, familial combined hyperlipidemia, abetalipoproteinemia, high-density lipoprotein (HDL) deficiency, and “other dyslipidemias” where the ambiguity of “primary hyperlipidemia” in the previous list remained. Such inconsistency between the lists creates substantial confusion in the transition of some cases and causes trouble in the continuation of medical care subsidization (Table 1, Table 2).
Table 2.The discrepancy in classification between the Specific Pediatric Chronic Diseases and the Adult Intractable Diseases
| The Adult Intractable Diseases (Assigned Number and Name of the Disease)
|
| 79 |
Familial hypercholesterolemia (homozygote) |
| 259 |
Lecithin cholesterol acyltransferase deficiency |
| 260 |
Sitosterolemia |
| 261 |
Tangier disease |
| 262 |
Primary hyperchylomicronemia |
| 263 |
Cerebrotendinous xanthomatosis |
| 264 |
Abetalipoproteinemia |
| 336 |
Familial Hypobetalipoproteinemia 1 (homozygote) |
| The Specific Pediatric Chronic Diseases (Assigned Number and Name of the Disease)
|
| 129 |
Primary hyperchylomicronemia |
| 130 |
Familial hypercholesterolemia (both homozygote and heterozygote) |
| 131 |
Familial combined hyperlipidemia |
| 132 |
Abetalipoproteinemia |
| 133 |
High-density lipoprotein deficiency |
| 134 |
Lipid metabolism disorders, in addition to those listed in 129 to 133 |
Medical expense subsidization is to be applied and reviewed by the prefectural government so that the decision sometimes fails to keep track of nationwide consistency. In fact, a few cases of such confusion have been reported in some areas. “The Committee on Primary Dyslipidemia under the Research Program on Rare and Intractable Disease of the Ministry of Health, Labour and Welfare of Japan “ (hereafter referred to as “this research group”) is currently working to resolve this problem on a priority basis.
II General Remarks: Issues in Transitional Medical Care of Primary Dyslipidemias in Japan
1. Medical Care Challenges during the Transition Period
What is required in transitional medicine for childhood-onset diseases that require lifelong treatment? First, patients who can be self-reliant are to be helped to acquire health literacy (the ability to obtain, understand, and make effective decisions about the information they need about their health) and develop self-management skills1). Ultimately, the patient must become an adult who takes responsibility for their health care. In the medical care transition of primary dyslipidemia, finding counterpart physicians for adults in some regions may be difficult. In case of difficulty in finding a counterpart, the primary care physician who treated the pediatric patient needs to continue the care by coordinating with the adult department.
Other issues in transitional medicine include poor adherence or drop-out, management during pregnancy and postpartum, and management of complications that are more pronounced in adulthood than in childhood. Since young patients leave home for higher-level education often coincides with the transition period, they are required to find a new appropriate physician in charge, and it is better to carry a medical card that indicates the details of their illnesses. It is also necessary to explain the genetic background through genetic counseling.
2. The goal of Transitional Medicine
The Japan Pediatric Society published “Recommendations for Transitional Care of Patients with Childhood-Onset Diseases” in 2013 2). The following four concepts were presented in this proposal. (1) It is essential that the patients themselves receive explanations and be able to make decisions or express their opinions according to their comprehension and judgment. (2) Regarding childhood-onset diseases in adulthood, it is essential to study the changing pathophysiology with age and to develop appropriate treatment methods. (3) At the same time, as pathological conditions change and personalities mature, these two providers are expected to provide seamless medical care during the transition from pediatric care to adult care. (4) Since the patient-parent-provider relationship changes as the patient’s personality matures, the healthcare system should be selected to correspond to this change and to match the individual’s disease and other characteristics.
Furthermore, in 2023, a new recommendation was released from the Japan Pediatric Society, “Recommendations to Promote Support for Transition to Adulthood for Patients with Childhood-Onset Chronic Diseases”3). These recommendations include the following: basic stances (respect for the right to self-determination, seamless medical care through medical coordination, team support by multiple professions, understanding of systems related to transitional care), support for transition (support for independence and self-reliance, preparation for transition, support for parents, continuation of medical care, support related to sex, pregnancy, and childbirth, support for social participation), support for transition to adulthood (patient acceptance, coordination for transition, timing of transition, medical summaries, response to emergency and hospitalization, follow-up after transition), system development and else (role of academic societies, financial support, transition support for patients with intellectual and developmental disabilities (neurodevelopmental disorders), research and feedback). Thus, the importance of the concept of transitional care is described, i.e., support for transition to adulthood from a broad perspective that covers not only medical care but also health and welfare.
As discussed below in each section, the following points are issues to address in primary dyslipidemia. (1) discrepancy in classification between the Specific Pediatric Chronic Diseases and Adult Intractable Diseases (Table 2), (2) inadequate adolescent diagnosis rates, (3) collaboration among specialist physicians, and (4) lack of understanding of the natural prognosis of rare diseases. From a healthcare aid perspective, designing a system that prevents dropouts, such as interrupted medical visits during the transition period, is necessary. In addition, disease awareness in the pediatric field is still insufficient. These issues should be addressed, and efforts should be made toward seamless transitional care for primary dyslipidemia.
3. Issues in the Japanese Healthcare System for Transitional Medicine of Primary Dyslipidemia
(1) Discrepancy in Classification between Specific Pediatric Chronic Diseases and Adult Intractable Diseases (Table 2)
As shown in Table 2, there is a discrepancy in classification between the diseases covered by the Specific Pediatric Chronic Diseases and the designated Adult Intractable Diseases. The adult list identifies eight specific genetic diseases, while the pediatric list uses less specific criteria, resulting in broader coverage of diseases by the latter. This means that the adult list cannot save substantial numbers of diseases covered in the pediatric list, and some patients may be terminated from the aid for expensive life-supporting treatment. This situation must be avoided, although there is no need for a consistent classification. Among those whom the adult list may not save, heterozygous FH is mostly treatable by conventional medications. Familial combined hyperlipidemia is not a strictly established clinical entity and is also perhaps conventionally treatable. “Other primary dyslipidemia” may include primary type III hyperlipoproteinemia and homozygous hypobetalipoproteinemia 1. The former is conventionally treatable, and the latter is included in the adult list. Apolipoprotein A-I deficiency is included in the “high-density lipoprotein deficiency” criteria in the pediatric list, but there are no matching criteria in the adult list. Therefore, this disease corresponds to this situation. This research group already and repeatedly filed this in a new application for designation as an Adult Intractable Disease.
(2) Insufficient Diagnosis Rate and Small Number of Beneficiaries
For the general pediatrician who is routinely involved in the treatment of many chronic and acute illnesses, making a diagnosis of primary dyslipidemia may be extraordinary. The low rate of diagnosis may have several backgrounds. Blood tests are less commonly performed in the pediatric field. Intractable diseases with complex lipid metabolism abnormalities often do not present a disease-specific phenotype in childhood. A lack of detailed awareness of rare diseases may exist even among lipid specialists, and a limited number of specialists may be available in a given region. Another factor that makes it challenging to increase the number of beneficiaries of the Specific Pediatric Chronic Diseases program may be the generous subsidies provided by local governments. In other words, there is no financial benefit to applying for the program since childhood medical care is completely free or partially paid for by the local government in Japan. Some of the severely affected children are recipients of subsidies under the Act on Welfare of Physically Disabled Persons. With this background, patients are not registered in the program for Specific Pediatric Chronic Diseases or Adult Intractable Diseases. This makes it difficult to estimate the nationwide number of patients suffering from these diseases.
The Ministry of Health, Labour, and Welfare’s perspective on transitional medicine aims for physicians who treat adult diseases, such as internists, to understand pediatric chronic diseases. Since dyslipidemia specialists are more in the field of internal medicine than in the field of pediatrics, reverse and bidirectional understanding is necessary for the diseases targeted discussed above. Two-way communication is still insufficient.
(3) Treatment System Issues
There is an uneven regional distribution of lipid specialists, so collaboration between pediatricians and lipidologists is not quickly established. In addition, cross-sectional efforts across academic societies are insufficient.
(4) Lack of Understanding of the Natural Prognosis of Rare Diseases
Since there was no long-term nationwide registry study of primary dyslipidemia conducted, the realistic prognosis of these diseases is not fully understood. A registry study of the (PROLIPID study, see below) finally started in 2015 4).
4. Efforts of the Primary Dyslipidemia Research Group, Unresolved Issues, and Future Vision
(1) Enforce Collaboration across Academic Groups throughout Japan; Establishment of a Transitional Medicine Workgroup
The mission of this research group includes designing a medical treatment system targeting intractable primary dyslipidemia and resolving remaining issues in transitional medicine. The group aims to propose a nationwide medical care system that patients in communities can access. In the care of primary dyslipidemia, there are many unresolved issues related to transitional care, as described above.
(2) Efforts to Eliminate Patients who are Unable to Transition to the Designated Adult Intractable Diseases
As noted above, there is a discrepancy in classification between the Specific Pediatric Chronic Diseases and the designated Adult Intractable Diseases. Therefore, affected children with the diseases listed in the Specific Pediatric Chronic Diseases but not in the designated Adult Intractable Diseases are disadvantaged during the transition period. Such diseases include FH heterozygotes, familial combined hyperlipidemia (FCHL), and apolipoprotein A-I deficiency. The ambiguous group of “other primary dyslipidemia” in the pediatric list may include primary type III hyperlipoproteinemia and familial hypo-beta lipoproteinemia 1 (homozygous).
FH heterozygotes are frequent, occurring in approximately 1 in 300 persons in the general population. Some patients are at high risk of developing coronary artery disease and require expensive treatments such as proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors and lipoprotein apheresis therapy. However, if all patients were to be designated as having Intractable Diseases, it might affect the sustainability of the current medical system for intractable diseases in Japan, so patients are treated by standard insurance after the age of 20. Some severe heterozygous FH patients are unable to start or have given up treatment due to the high medical costs. Therefore, taking necessary measures to ensure these patients are not disadvantaged is essential.
Because FCHL is not a monogenic disease and patients present with a wide variety of dyslipidemias, it is often difficult to determine whether a patient has FCHL or secondary dyslipidemia. Although diagnostic criteria for FCHL exist, the concept of the disease is still under debate. As mentioned above, as with less severe FH heterozygotes, blood lipid levels can often be managed with relatively inexpensive conventional treatment in many patients, so currently, new applications are not being submitted as a designated Adult Intractable Disease. The number of children with FCHL registered for the Specific Pediatric Chronic Diseases is very small, so only a small number of patients can be treated during the transition period if nay5).
As for type III hyperlipoproteinemia, the causative gene is known, but as with the less severe FH heterozygotes, blood lipid levels can often be managed with relatively inexpensive conventional treatment in most patients, so currently, no new applications are being filed as a designated Adult Intractable Disease.
Apolipoprotein A-I deficiency is included in HDL deficiency in Specific Pediatric Chronic Diseases, and to date, there is no established treatment for this disease, and premature coronary artery disease is a life prognostic factor. We believe this was omitted from the application list during organizing the designated Intractable Diseases for enforcing the Intractable Disease Law (Act on Medical Care for Patients with Intractable Diseases) in 2015. This research group has applied to designate the disease as a new intractable disease. The application has not been approved for designation as an Intractable Disease so far as 2023.
As for familial hypobetalipoproteinemia1 (homozygous), a disease that presents the same severe phenotype as abetalipoproteinemia (MTP deficiency), this research group applied for the designation as a new Intractable Disease, and it was designated as the 336th intractable disease in 2020.
III. Overview of Each Primary Dyslipidemia and Transitional Medicine
In this chapter, the 12 primary dyslipidemias are divided based on the consistency in classification between the Specific Pediatric Chronic Diseases and the designated Adult Intractable Diseases. We will then describe each disease in outline from the perspective of transitional care.
1. Diseases Defined as both the Specific Pediatric Chronic Diseases and the Designated Adult Intractable Diseases
(1) Sitosterolemia
Sitosterolemia is an inherited dyslipidemia with an autosomal recessive manner, in which decreased excretion of sitosterol, a type of phytosterol found in fruits and vegetables, results in accumulation of sitosterol and cholesterol in the blood or tissues, causing xanthomas and premature coronary artery disease and other clinical manifestations6). Abnormal red blood cells, hemolytic attacks, thrombocytopenia, and arthritis may also be seen. As for the cause of sitosterolemia, genetic mutations in the ATP-binding cassette transporter (ABC) G5/8 (ABCG5/ABCG8) are involved in the pathogenesis of the disease. Sterols in the diet are absorbed by the sterol transport protein Niemann-Pick C1 like 1 (NPC1L1) in the small intestine. Cholesterol is esterified in the small intestinal epithelium and used to form chylomicrons (CMs), while unused plant sterols and excess cholesterol are excreted into the intestinal tract via ABCG5/8. In sitosterolemia, functional abnormalities associated with mutations in the ABCG5/8 gene result in impaired excretion of phytosterols and cholesterol, causing them to accumulate in the body. Accumulated phytosterols (mostly sitosterol) and cholesterol are deposited in tissues such as skin and tendons, forming xanthomas, accumulating in blood vessel walls, and forming atherosclerotic plaques. Dietary therapy is based on limiting vegetable oils, nuts, and cereals and avoiding foods high in plant sterols (corn, sesame, peanuts, soybeans, rapeseed oil, sesame oil, rice oil, margarine, nuts, avocados, chocolate, etc.) and shellfish as much as possible. Still, other vegetables and fruits can be consumed. Cholesterol restriction should be less than 200 mg per day7). However, there are some cases in which dietary treatment is not effective, and drug therapy such as ezetimibe or cholestimide (resin) is often required. The diagnosis may be triggered by marked high LDL cholesterol levels and cutaneous xanthomas during infancy, especially with breastfeeding. With weaning, LDL cholesterol levels improve markedly, and the xanthomas often disappear spontaneously, and this phenomenon is a trigger for suspicion of the disease. There are no reports of retardation of development or growth in childhood or subsequent reproductive abnormalities. Although the name of the disease, sitosterolemia, is not included in the list of Specific Pediatric Chronic Diseases, it could be registered as “lipid metabolism disorders in addition to those listed in 129 to 133”.
(2) Primary Hyperchylomicronemia
Chylomicrons are lipoproteins that transport dietary lipids and are secreted from the small intestine and then metabolized by lipoprotein lipase (LPL) in the blood, which hydrolyzes the triglycerides (TG) in the chylomicrons. Primary hyperchylomicronemia is a disease in which chylomicron levels in the blood are markedly increased due to genetic mutations or autoantibodies to molecules involved in triglyceride hydrolysis. LPL-related molecules include lipase maturation factor 1 (LMF1), glycosylphosphatidylinositol-anchored high-density lipoprotein binding protein 1 (GPIHBP1), apolipoprotein A5 (ApoA5), and ApoC2 8). Even LPL deficiency, considered the most frequent form of the disease, occurs in only about 1 in 1 million people, which is extremely rare. Typical cases begin in childhood and present with symptoms such as abdominal pain associated with pancreatitis, as well as eruptive xanthomas, lipemia retinalis, and hepatosplenomegaly. Some cases have adult onset, and others are exacerbated only when there is a burden, such as pregnancy. In general, the higher the blood TG level, the higher the risk of pancreatitis, but the risk is even higher for primary hyperchylomicronemia due to genetic factors at the same TG level9).
Since there is no curative treatment and existing TG lowering therapy has limited efficacy, restriction of dietary fat intake (15-20 g or less per day (10-15% or less of daily calories)) is the cornerstone of treatment10-14). Under fat restriction, adequate intake of essential fatty acids (EFA; 2-4% of daily calories) and fat-soluble vitamins (A, D, E, and K) should be ensured to avoid deficiency. Consider using MCTs and milk or skimmed milk powder containing MCTs to avoid nutritional disturbances. However, be aware that some reports suggest liver injury or cirrhosis due to long-term use of MCTs10, 11). Excessive intake of carbohydrates and alcohol intake are also factors that can exacerbate serum TG levels. Fat restriction in childhood is significant, and the earlier the diet is introduced, the better adherence to fat restriction throughout life11, 13). In transitional care, it is essential to provide support that considers the emotional, psychological, social, and financial burdens of changing life events, such as schooling, exams, higher education, employment, and marriage15, 16). Since pregnancy is a trigger for sudden exacerbation of hyperchylomicronemia and acute pancreatitis17), preconception counseling is recommended. More severe cases require more stringent restrictions on fat intake, and it is helpful to note that more stringent fat intake restrictions (<2 g/day) have been reported to be beneficial in severe cases of pregnancy10, 11).
(3) Cerebrotendinous Xanthomatosis (CTX)
CTX is an autosomal recessive lipid storage disorder caused by mutations in the CYP27A1 gene, which encodes the mitochondrial enzyme sterol 27-hydroxylase. In Japan, only approximately ten patients are newly diagnosed annually18), but since genetic epidemiological studies estimate the frequency of CTX to be 1 in 64,267-64,712 East Asians19), it is likely that many patients remain undiagnosed. Generally, routine lipid tests are normal. Serum total cholesterol is normal to mildly decreased. Serum cholestanol levels, an uninsured test in Japan, are markedly increased, but the relationship to the extent of clinical symptoms is unclear20).
Clinical phenotypes can be broadly classified into the following three categories20). The classical form presents with progressive neuropathy (such as intellectual impairment, pyramidal signs, and cerebellar signs), Achilles tendon xanthomas, juvenile cataracts, and premature cardiovascular disease. Patients with the spinal form exhibit clinical symptoms and signs related to the involvement of the spinal cord’s corticospinal tracts and dorsal columns without intellectual impairment, cerebellar signs, or peripheral neuropathy at the time of the presentation of spinal cord syndrome. Patients with the non-neurological form have no neuropsychiatric manifestations.
The possibility of CTX should be suspected in neonatal cholestasis. Neonatal mass screening using bile acid markers is also reported21). Many patients also have diarrhea that develops in infancy and childhood21). Since the symptoms in childhood are nonspecific, a score to suspect CTX (suspicion index score) has been devised22). On the other hand, neurological symptoms, as well as cataracts, have been observed since childhood.
Chenodeoxycholic acid is used as a therapeutic agent23) and markedly reduces serum cholestanol levels. Although chenodeoxycholic acid improves diarrhea early in the disease, the neurological symptoms are irreversible. However, it is important to note that early diagnosis of CTX is expected to prevent neurologic complications21). Lipoprotein apheresis (not covered by insurance) may be considered an option if response to therapy is poor24). As for dietary therapy, nothing fundamentally improves the condition, but diet is essential to prevent atherosclerosis risk.
(4) Abetalipoproteinemia (ABL)/Familial Hypo-Beta-Lipoproteinemia (FHBL) 1 (Homozygote)
Abetalipoproteinemia (ABL) is an autosomal recessive form of inherited disease caused by an abnormality in the MTTP gene encoding microsomal triglyceride transfer protein25-27). On the other hand, familial hypobetalipoproteinemia (FHBL) 1 (homozygote) is an autosomal dominant form of inherited disease caused by an abnormality in the APOB gene, which encodes apolipoprotein B26, 28, 29). The frequency of both diseases is reported to be less than 1 in 1 million, which is extremely rare. While ABL is listed as one of the Specific Pediatric Chronic Diseases, FHBL1 homozygotes can be registered as “lipid metabolism disorders in addition to those listed from 129 to 133”. Defects in MTTP and apolipoprotein B impair synthesis and secretion of VLDL and chylomicrons, resulting in hypolipidemia (serum LDL-C <15 mg/dL or serum apolipoprotein B <15 mg/dL) and impaired absorption of lipids and fat-soluble vitamins, which in turn leads to nutritional disorders and various organ damage27). Symptoms include chronic diarrhea and growth retardation due to impaired fat absorption, ocular symptoms (loss of night vision and color vision, retinitis pigmentosa, vision loss, etc.), and neurological disorders (spinocerebellar degeneration, ataxia, peripheral neuropathy, etc.). In severe cases, vomiting, abdominal distention, diarrhea due to fat malabsorption, and growth retardation due to nutritional disorders begin after the onset of breastfeeding, neurological disorders appear between the ages of 2 and 10, and ocular symptoms appear by puberty. Because of the wide variety of symptoms, it is important to collaborate with other medical departments.
No significant differences in clinical symptoms or laboratory findings exist between ABL and FHBL1 homozygotes, but a family history helps differentiate the two. ABL is inherited in an autosomal recessive manner, which means that first-degree relatives do not have hypolipidemia. In contrast, FHBL1 homozygotes are inherited as an autosomal co-dominant trait, which means that first-degree relatives (FHBL1 heterozygotes) have moderate hypolipidemia. The final diagnosis should be made by genetic testing. In making the diagnosis, follow the diagnostic criteria for ABL/FHBL1 homozygosity27, 28) and differentiate other hypolipidemias30).
Although there is no curative treatment, early diagnosis, early initiation of treatment (diet and vitamin replacement therapy), and continued lifelong medical guidance are essential to reduce symptoms and improve prognosis25-34). ABL and FHBL1 homozygotes are not significantly different in treatment strategy. Restriction of fat intake is necessary to prevent diarrhea, fatty stools, and secondary malabsorption resulting from fat malabsorption. The use of MCTs may also be considered to avoid the resulting caloric deficit. However, be aware of liver dysfunction and cirrhosis caused by long-term administration of MCTs31, 32). High-dose vitamin replacement therapy may help alleviate symptoms and improve the prognosis for neurological disorders, mainly caused by vitamin E deficiency, and ocular symptoms, which are thought to be caused by vitamin E and A deficiency. However, women who are pregnant or may become pregnant require volume adjustment to avoid vitamin A overload. Vitamin D and K supplementation may also be necessary. Vitamin B12, vitamin B6, iron, and folic acid supplementation may also be required. See references 25)-28) and 33) for recommended fat intake and vitamin supplementation in childhood and adulthood. Since this is a rare disease, further accumulating cases and evidence on appropriate dosages is desirable.
Follow-up during the transition period should include periodic evaluation of nutritional status, testing of blood levels of fat-soluble vitamins, and evaluation of symptoms attributable to fat-soluble vitamin deficiency. Ophthalmologic abnormalities, neurologic abnormalities, fatty liver, osteoporosis, myocardial disorders, and anemia are regularly evaluated (see references 27), and 28) for a detailed list). Early treatment is essential because the later treatment is initiated, the less likely symptoms will recover.
2. Specific Pediatric Chronic Diseases for which Medical Subsidies are no Longer Available or Limited when the Patient Reaches Adulthood
(1) Familial Hypercholesterolemia (FH)
FH is a disease caused by gene mutations encoding low-density lipoprotein (LDL) receptors and related proteins35). Mutations in the LDL receptor gene (LDLR) are frequent36), but there are also cases caused by gain-of-function mutations in the PCSK9 gene, which is involved in LDL receptor degradation, and by abnormalities in the apolipoprotein B gene (APOB), which is extremely infrequent in Japan37). Although FH is usually inherited in an autosomal dominant form, autosomal recessive hypercholesterolemia (ARH), which is extremely rare, has also been reported37). FH can be homozygous for a genetic abnormality inherited from both parents and a single parent with double heterozygous.
While both homozygotes and heterozygotes for FH are Specific Pediatric Chronic Diseases, only homozygotes can be designated as the designated Adult Intractable Diseases38, 39). Thus, in heterozygotes, medical subsidies received in childhood are discontinued when the patient reaches adulthood. The reason for this is believed to be that if FH heterozygotes, which have the highest frequency among hereditary metabolic diseases (1 in 300), are subsidized as an adult-designated incurable disease, the medical cost to be paid by the government would be enormous. There is a fear that the intractable disease medical care system would be unable to continue. On the other hand, the number of registered patients as FH in Specific Pediatric Chronic Diseases was 151 in 2018 and 31 in 2019, a marked decrease5). The number of children treated without registration is increasing because each municipality subsidizes medical expenses, so it is difficult to determine the number of patients nationwide from the number of registrations.
In FH, serum LDL-C levels are high from birth, resulting in rapid progression of atherosclerosis, and untreated patients are at increased risk of developing angina pectoris and myocardial infarction at a young age. On the other hand, if appropriate therapeutic intervention is provided from childhood, the prognosis is similar to that of non-FH patients. In other words, FH is a critical disease in pediatrics regarding the need for early diagnosis and treatment35).
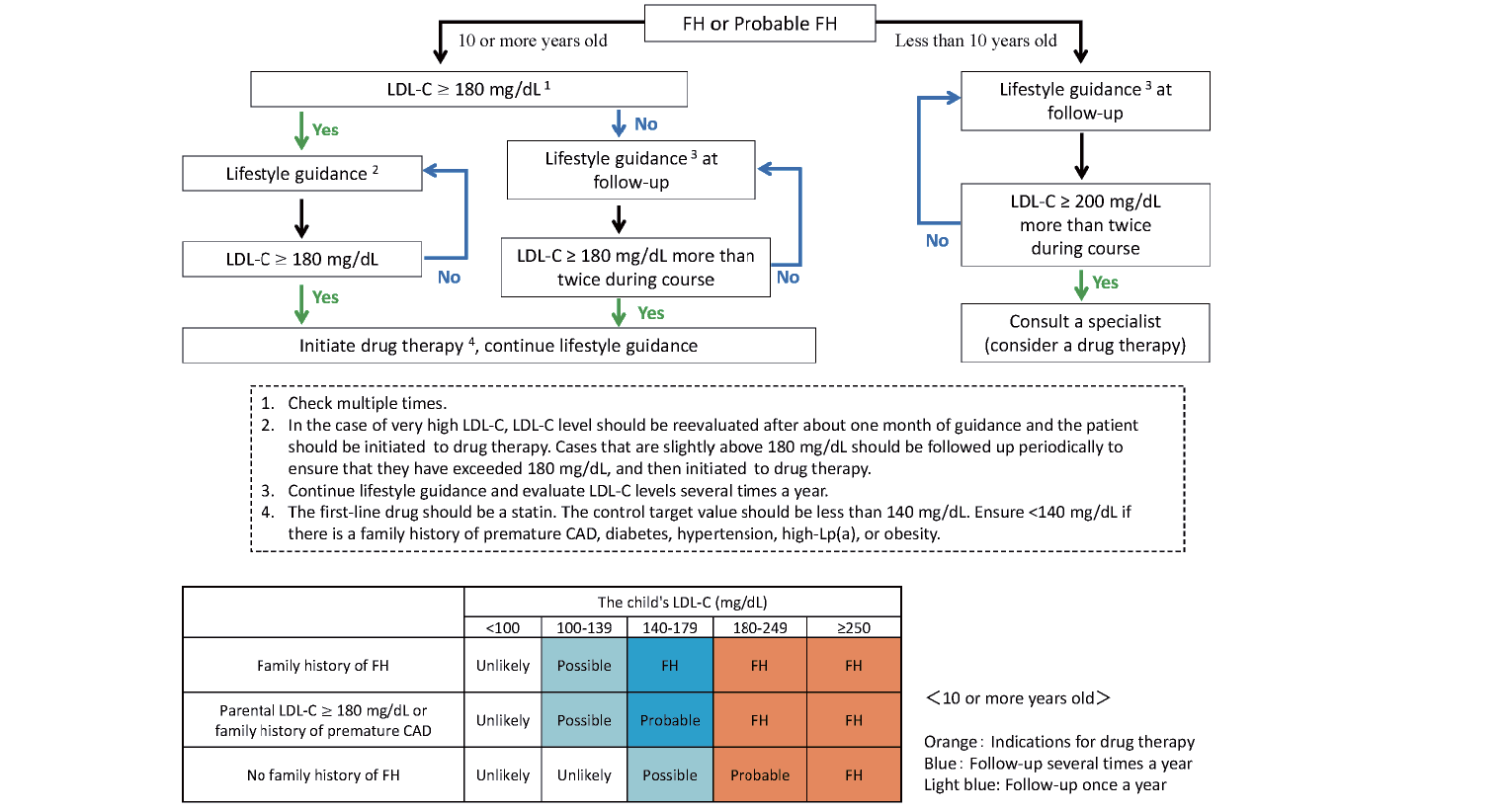
The diagnostic criteria for FH in Japan differ slightly between adults (15 years and older)40) and children. As xanthomas and Achilles tendon thickening are usually absent in pediatric patients with heterozygous FH, the diagnosis is based on high LDL-C levels (>140 mg/dL) and a family history of FH. Notably, the presence of skin/tendon xanthomas suggests severe disease and a suspicious finding for homozygous FH35). The diagnostic criteria35), newly revised in 2022 (Table 3), are designed to increase diagnostic sensitivity over the 2017 pediatric FH diagnostic criteria41) by establishing a “probable FH” category. In making the diagnosis, multiple tests should be performed, considering that LDL-C levels fluctuate during puberty. Treatment of FH heterozygotes begins with lifestyle modification, including diet and weight. Pharmacological therapy should be considered for patients of both sexes aged ten years and older with persistent LDL-C >180 mg/dL and those younger than ten years of age with persistent LDL-C >200 mg/dL. First-line therapy should be a statin with a target LDL-C of 140 mg/dL. If statins alone are insufficient, consider concomitant use with other drugs. Although there have been no reports of serious adverse effects of statin administration in childhood, growth and development should be monitored carefully35). In FH homozygotes, preparation for induction of lipoprotein apheresis should be done while administering drugs such as high-intensity statins. The initiation of apheresis treatment is often required before the start of elementary school35). Details of treatment strategies are discussed below.
Table 3.Diagnostic criteria for pediatric FH (under the age of 15)
| 1. Hyper-LDL cholesterolemia (untreated LDL-C level ≥ 140 mg/dL, confirmed multiple times) |
| 2. Family history of FH (Parents or siblings) |
| 3. Parental LDL-C ≥ 180 mg/dL or family history of premature coronary artery disease (Grandparent or parent) |
| After ruling out other primary and secondary Hyper-LDL cholesterolemia, |
-
● Diagnose FH with items 1 and 2.
|
-
● Diagnose probable FH with items 1 and 3. If the individual‘s LDL-C is 180 mg/dL or higher, diagnose FH.
|
-
● Even if only item 1 is used, ≥ 250 mg/dL is diagnosed with FH, and ≥ 180 mg/dL is diagnosed with probable FH.
|
-
・Differentiate HoFH when LDL-C is ≥ 250 mg/dL or xanthomas are present.
|
-
・Diagnose FH if the individual has a pathogenic gene mutation for FH.
|
| If a parent, a brother, or a sister is found to have a pathogenic gene mutation for FH, that is considered to be the family history of FH (item 2). |
-
・Premature coronary artery disease is defined as coronary artery disease occurring at less than 55 years of age in men and less than 65 years of age in women.
|
-
・Probable FH cases require further scrutiny and lipid-lowering therapy.
|
HDL deficiency, one of the terms defined for Specific Pediatric Chronic Diseases, includes lecithin cholesterol acyltransferase (LCAT) deficiency, Tangier disease, and apolipoprotein A-I deficiency. Among these three diseases, LCAT deficiency and Tangier disease are designated as Intractable Diseases for Adults, but apolipoprotein A-I deficiency is not. This means that medical subsidies for patients with apolipoprotein A-I deficiency who have reached adulthood will be discontinued.
(i) LCAT Deficiency
LCAT deficiency is included in “HDL deficiency” in the Specific Pediatric Chronic Diseases system and would be considered one of them. LCAT deficiency is an inherited disease with an autosomal recessive form caused by mutations in the LCAT gene, which encodes LCAT. It is extremely rare, with no racial variation. It is estimated to account for 2-9% of cases of markedly low HDL-C levels42). In Specific Pediatric Chronic Diseases, it is registered as “HDL deficiency,” but there were 0 patients in 2019 5). LCAT deficiency is divided into two phenotypes: familial LCAT deficiency (FLD) and fish eye disease (FED)42-44). The frequency of FED is reported to be approximately one-third that of FLD42, 44). Both have corneal opacity and markedly low HDL-C (typically <10 mg/dL). Phenotypic variability, even in patients with the same genetic mutation, suggests a role other than genetic factor45).
It should be noted that some patients with LCAT deficiency have serum HDL-C levels >20 mg/dL43, 46). HDL-C is almost always measured by direct methods, but reactivity to HDL with abnormal composition varies with the reagent. Many cases present with high TG levels up to 400-500 mg/dL. LDL-C is lower in FLD than in FED. This is because LCAT activity for endogenous lipoproteins is almost defective in FLD but is reduced to the lower end of the reference range in FED. Half of FLD patients have proteinuria or albuminuria, and approximately 40% have impaired renal function; more than 70% of FLD patients have hemolytic anemia. These conditions are less common in FED, but on the contrary, premature coronary artery disease is present in nearly 30% of patients (rarely in FLD). Even though corneal opacity and proteinuria are present from childhood in FLD, the median age at diagnosis is around 30 years, with few pediatric cases44). At present, there is no valid treatment. Administration of recombinant LCAT and small molecule LCAT activators have been investigated in preclinical studies45). In Japan, a clinical trial of gene therapy using LCAT transgenic preadipocyte transplantation is underway.
As mentioned above, there are few cases diagnosed in childhood. Since corneal opacity can be noticed as early as childhood, measuring HDL-C in children with hypocholesterolemia and corneal opacity may lead to early detection. Lipid testing, including HDL-C, is also recommended when proteinuria is present. As for dietary therapy, it has been reported that a low-fat diet reduces proteinuria and delays the progression of renal dysfunction47-50).
(ii) Tangier Disease
Tangier disease is included in the “HDL deficiency” category in the Specific Pediatric Chronic Diseases and would be considered one of them. Tangier disease is a genetic disorder with an autosomal recessive form and is caused by mutations in the ATP-binding cassette transporter A1 gene (ABCA1)51). According to genetic epidemiological studies, the frequency of loss-of-function variants in ABCA1 is approximately 1/400, so Tangier disease is thought to exist in 1 in 640,000 people worldwide. Still, the number of patients registered as “HDL deficiency” as a Specific Pediatric Chronic Disease is 0 5) in 2019, suggesting that many cases are undiagnosed. Blood lipid tests are characterized by extremely low HDL-C levels and lower apolipoprotein A-I levels than in LCAT deficiency52). Some cases may present with elevated TG levels. The phenotype includes orange tonsillar enlargement, hepatosplenomegaly, corneal opacities and peripheral neurological symptoms, abnormal glucose tolerance, premature coronary artery disease, mild thrombocytopenia, and hemolytic anemia. Currently, there is no effective therapy. To prevent coronary artery disease, which determines life prognosis, it is essential to control lipids other than HDL-C (namely LDL-C and triglycerides), manage risk factors such as hypertension and smoking, pay attention to abnormal glucose tolerance, and prevent the onset of diabetes52, 53). In childhood, phenotypes other than orange tonsils and hepatosplenomegaly are scarce, and it is believed that this disease is easily overlooked because blood samples for lipids are rarely taken during pediatric visits. It is recommended that HDL-C levels be measured in cases of hypolipidemia and orange tonsils, and disease awareness should be raised. Early detection may improve patient prognosis by allowing management of coronary risk factors other than HDL-C.
(iii) Apolipoprotein A-I Deficiency
Apolipoprotein A-I deficiency is included in the “HDL deficiency” category in the Specific Pediatric Chronic Diseases and is considered one of them, but it is not included in the designated Intractable Diseases for Adults and may not continue to receive medical expense assistance at present. Apolipoprotein A-I deficiency is an autosomal codominant inherited disorder caused by mutations in the APOA1 gene (APOA1) encoding apolipoprotein A-I protein. It is difficult to determine the frequency and number of patients in Japan. Blood lipid tests show extremely low HDL-C levels, and apolipoprotein A-I levels are not detected in homozygotes or compound heterozygotes. Heterozygotes also have low HDL-C levels, but apolipoprotein would be measurable. Phenotypes of homozygous or compound heterozygous patients include corneal opacities, xanthomas, and premature coronary artery disease. Some mutations cause familial amyloidosis. Currently, there is no valid therapy, and management of risk factors other than HDL-C is essential to prevent coronary artery disease, which determines life prognosis. The disease may be easily overlooked in childhood due to the lack of phenotype and infrequent blood sampling. Measuring HDL-C levels in cases of hypolipidemia is recommended, and disease awareness should be raised. Early detection may improve patient prognosis by allowing management of coronary risk factors other than HDL-C53).
3. Diseases Designated under the Specific Pediatric Chronic Diseases but not Designated as the Adult Intractable Diseases
(1) Type III Hyperlipoproteinemia
Type III hyperlipoproteinemia is an inherited lipid disorder caused by an abnormality of apolipoprotein E. Apolipoprotein E has isoforms E2 and E4 in addition to wild-type E3, and E2/E2 is the predominant form in type III hyperlipoproteinemia, with rare reports of E1 and E deficiency. Remnant lipoproteins accumulate in the blood mainly due to dysfunction of apolipoprotein E, which acts as a ligand for lipoprotein receptors expressed in the liver. E2/E2 alone does not cause marked dyslipidemia, but it is manifested by complications such as obesity, diabetes mellitus, and hypothyroidism. The frequency of E2/E2 in Japan is estimated to be approximately 0.2%, but only about 10% of these patients develop the disease. A significant phenotype is a high risk of developing premature coronary artery disease54), and remnants may be deposited in the tissues and present as palmar linear xanthomas or cutaneous nodular xanthomas. Laboratory findings include high levels of both total cholesterol and TG, with TG levels sometimes exceeding 1,000 mg/dL. The proof of broad β pattern by agarose electrophoresis of lipoproteins and high apolipoprotein E levels helps diagnose. Treatment includes diet and other lifestyle interventions, as well as therapy for comorbid obesity, diabetes, and thyroid disease. Lipid-lowering drugs such as fibrates and statins are effective. Although childhood diagnosis is difficult, measuring lipoprotein electrophoresis and apolipoproteins in patients with elevated TC and TG levels is recommended. If the disease is detected at an early stage, lifestyle modification and, if necessary, medication can be given to improve the prognosis.
(2) Familial Combined Hyperlipidemia
Familial combined hyperlipidemia (FCHL) was once thought to be a single-gene disorder with autosomal dominant inheritance, which is common in patients with myocardial infarction. However, it is currently considered a multifactorial disease influenced by some genetic factors combined with factors such as overnutrition, obesity, and lack of physical activity. In the past, screening of 56,181 children was conducted by this study group from 1990 to 1999, and it was reported that a significant number of children presenting with type IIb have FCHL55, 56).
Within the same patient, serum lipids profile can fluctuate, with high levels of both LDL cholesterol and TG or only one of them. Increased apoprotein B and smaller LDL particle size are observed. A family history of dyslipidemia or coronary artery disease is noted, but no Achilles tendon thickening or other xanthomas are present. Treatment begins with lifestyle intervention, and if the patient does not respond, statins, fibrates, or ezetimibe are prescribed.
IV Efforts to Improve Transitional Medicine for Primary Dyslipidemia
1. Efforts to Improve Diagnosis Rates
(1) Original Website of the Research Group for the Primary Dyslipidemia
One of the difficulties in diagnosing rare diseases is the inaccessibility of accurate information regarding diagnostic criteria and treatment strategies. To address this point, the Primary Dyslipidemia Research Group has released the official website (https://nanbyo-lipid.com/). On the website, information on each disease is available for patients and their families, including the pathophysiology of the disease, lifestyle guidance, and drug treatment, as well as information for healthcare professionals, including general physicians. The website also has an inquiry form for healthcare professionals and patients and offers consultations on everything from disease diagnosis and treatment to matters related to daily life.
(2) Guidelines for the Diagnosis and Treatment of Pediatric Familial Hypercholesterolemia 2022
In Japan, “The Guidance for Pediatric Familial Hypercholesterolemia 2017” was released in 2017 as a guideline for treating pediatric FH41). Five years later, in 2022, a revised version of “Guidelines for the Diagnosis and Treatment of Pediatric Familial Hypercholesterolemia 2022” was published35).
Diagnosis of FH is based on high LDL-C levels (>140 mg/dL) and family history of FH. Current diagnostic criteria for pediatric FH are shown in Table 3.
Since the publication of the practice guide in 2017, the diagnosis rate of pediatric FH seems to have improved. However, the diagnostic criteria published in 2017 (high LDL-C levels + family history of FH or premature coronary artery disease) did not lead to a diagnosis of FH in some cases due to unknown family history. As a result, it was reported that many cases were overlooked from the diagnosis, especially in pediatric lifestyle-related disease checkups57). Therefore, in the new diagnostic criteria (Table 3), “probable FH” was established to reduce missed cases. In addition, the diagnosis of FH is made when a pathogenic mutation is identified in the FH-related genes, and a family history of FH is added when a pathogenic gene mutation is found in a parent or a sibling. Furthermore, even if there is no apparent family history of FH, it can be evaluated by high serum LDL-C. A flowchart was also created to make the diagnosis easy to understand (Fig.2)35). If FH cannot be ruled out, the patient is asked to continue to see the doctor, including performing multiple blood tests. We believe that reliable diagnosis and therapeutic intervention will prevent atherosclerotic diseases in the future.

Until recently, family screening (cascade screening) has been the recommended screening method for FH in Japan. On the other hand, Kagawa Prefecture is actively promoting pediatric FH screening using the Pediatric Lifestyle Related Disease Prevention Health Checkup. As a result, significant progress is being made in improving the diagnosis rate of pediatric FH and reverse cascade screening of their parents57). In addition, the University of Tokyo has been implementing LDL-C screening for young adults using regular health checkups for new students. It has also achieved significant results in the early diagnosis of FH in adolescents.
(4) Collaboration among Academic Societies and Disease Awareness
Patients with primary dyslipidemia present with a variety of symptoms and physical findings other than blood lipid abnormalities, so they are sometimes diagnosed by physicians who are not lipidologists. However, diagnosis and treatment are delayed without knowledge of disease-specific symptoms and findings other than lipid abnormalities. Therefore, we believe it is crucial to raise awareness not only among related academic societies but also among a wide range of medical departments and to have interventions by multidisciplinary professionals other than physicians. Table 4 shows the symptoms and findings of each disease and the departments where awareness-raising is desirable.
Table 4.Symptoms and physical findings of intractable primary dyslipidemia and related medical departments
| Disease |
Symptoms and Physical Findings |
Related Medical Departments |
| Familial hypercholesterolemia (homozygote) |
Skin xanthoma, corneal arcus, Achilles tendon thickening, Achilles tendon pain, premature atherosclerosis |
Dermatology, Ophthalmology, Orthopedics, Cardiology |
| Lecithin cholesterol acyltransferase deficiency |
corneal opacity, renal dysfunction |
Ophthalmology, Nephrology |
| Sitosterolemia |
Skin xanthoma, Arthralgia, premature atherosclerosis |
Dermatology, Orthopedics, Cardiology |
| Tangier disease |
Orange-colored pharyngeal tonsils, hepatosplenomegaly, corneal opacity, Peripheral neuropathy, premature atherosclerosis |
Otorhinolaryngology, Hepato- gastroenterology, Ophthalmology, Neurology, Cardiology |
| Primary hyperchylomicronemia |
Lipemia retinalis, acute pancreatitis eruptive xanthoma, Exacerbations due to pregnancy |
Ophthalmology, Hepato-gastroenterology, Dermatology, Obstetrics and Gynecology |
| Cerebrotendinous xanthomatosis |
progressive neuropathy, juvenile cataracts, refractory diarrhea, cholestasis |
Neurology, Ophthalmology, Hepato- gastroenterology |
| Abetalipoproteinemia/ Familial Hypobetalipoproteinemia 1 (homozygote) |
diarrhea, fatty liver, night blindness, retinitis pigmentosa progressive neuropathy |
Hepato-gastroenterology Ophthalmology, Neurology |
The Prospective registry study of primary dyslipidemia (PROLIPID) study aims to clarify the natural history, medical care, and treatment of primary dyslipidemia4). Enrollment of patients with FH, type III hyperlipoproteinemia, and primary hyperchylomicronemia began in 2015, followed by enrollment of patients with sitosterolemia and CTX in 2019, and LCAT deficiency, Tangier disease, and ABL in 2020. The registry has started enrolling patients with FHBL1 homozygotes in 2022. Using the case data on primary dyslipidemia accumulated in this registry, we plan to elucidate each disease’s characteristic symptoms and findings, actual treatment status, and prognosis. By clarifying these data, we aim to reduce patient anxiety, revise medical guidelines, and improve patient prognosis.
Research can be conducted using the national database for Specific Pediatric Chronic Diseases and the designated Intractable Diseases for Adults. However, using the database for diseases with fewer than a certain number of patients is impossible because of the risk of personal identification in rare diseases. Recently, we reported on the actual practice of FH homozygotes in Japan using the national database58).
2. Lipid-Lowering Treatment for Pediatric FH
Lipid-lowering drug therapy for children should be judged on an individual basis. Among primary dyslipidemia, FH is a disease for which initiation of drug therapy in childhood is necessary and effective. FH is a high-risk condition for atherosclerotic disease, and “Guidelines for the Diagnosis and Treatment of Pediatric Familial Hypercholesterolemia 2022” recommends early initiation of treatment. The first-line drug is statin, which is also the case in Europe and the United States. Fig.3 shows the treatment chart for pediatric heterozygous FH, and Fig.4 for pediatric homozygous FH41).
Many lipid-lowering therapies used in adults are not approved for use in the pediatric setting in Japan. For instance, among the six statins approved in Japan, only pitavastatin is currently indicated for use in pediatric FH patients (pediatric indication has been obtained at age ten years and older). Many other countries have approved statins for pediatric use; e.g., in the U.S., pravastatin is approved for use at the age of eight years, and other statins are approved for use at the age of ten years41). Efficacy and safety of statins are being reported59).
Small intestinal cholesterol transporter inhibitors (ezetimibe) and resins (cholestimide and cholestyramine) have been used in pediatric patients for some time, but they are not approved for pediatric use. However, it is well known in Japan and overseas that combining statins and ezetimibe can effectively lower LDL-C in pediatric patients41). In addition, PCSK9 inhibitors (evolocumab and inclisiran) are not approved for use in children in Japan. Microsomal triglyceride transfer protein (MTP) inhibitor (lomitapide mesylate) has been approved only for FH homozygotes and has relatively many adverse effects. Therefore, these drugs are still used only in exceptional cases in children.
Newer agents are being developed. For example, a monoclonal antibody for angiopoietin-like protein 3 (evinacumab) was approved in the United States in 2021 to treat FH homozygotes. It is expected to be approved in Japan in 2024, and pediatric trials are underway.
3. Genetic Testing and Genetic Counseling
Among the primary dyslipidemias covered in this paper, the causative genes for all except FCHL have been identified, and genetic testing is practical. In addition, FH, primary hyperchylomicronemia, ABL, FHBL1 homozygous, and Tangier disease were covered by insurance in 2022. Although there are still many misconceptions about genetic testing for children, genetic testing for the diagnosis of symptomatic patients can be considered. On the other hand, the diagnosis of whether an asymptomatic child is a carrier or not, or pre-onset genetic testing for diseases that develop after adulthood, should be postponed until the child has reached adulthood and can make their own decision. Testing should not be conducted on behalf of parents or others (Guidelines for Genetic Testing and Diagnosis in Health Care, revised March 2022). Genetic counseling should also be provided during genetic testing and diagnosis. Genetic counseling provides information and psychological and social support so that patients can make their own choices. Therefore, a physician with extensive experience in treating the disease and a person skilled in genetic counseling should work together to provide genetic counseling. From this perspective, genetic information must be appropriately shared and stored over time, with due consideration to protect patient privacy and avoid unfair discrimination. In principle, the results of genetic tests and the content of genetic counseling should be recorded in the medical record as well as other medical information.
4. Efforts to Reduce Patient Anxiety and Improve Quality of Life
The research group supports the activities of the Patients’ Association for Intractable Familial Hypercholesterolemia. For example, a consulting physician who is also a research group member attends the general meeting of the patients’ association, provides direct consultation to patients, and lectures on recent advances in treatment and lifestyle interventions. The “Meeting of Hypercholesterolemia Patients,” jointly organized by the Patients’ Association and the research group, has been held four times to date. It included expert lectures and group discussions where patients could ask questions about things they usually wonder about. Sharing questions can help reduce anxiety because it helps patients realize that other patients have the same concerns. Also, the “Meeting of Sitosterolemia Patients” was held twice, where expert team members provided direct counseling to patients and education on the daily diet.
Funding
This study was supported by a grant from the Labor and Welfare Sciences Research Grant for Research on Rare and Intractable Diseases (21FC1009).
Conflict of Interest
Masatsune Ogura received lecture fees from Amgen and Kowa. Sachiko Okazaki and Hiroaki Okazaki received scholarships from Minophagen Pharmaceuticals and Kowa. Hayato Tada, Kazushige Dobashi, Kimitoshi Nakamura, Keiji Matsunaga, Takashi Miida, Tetsuo Minamino, and Shinji Yokoyama have nothing to disclose. Mariko Harada-Shiba holds stocks in Liid Pharmaceuticals and received payments or honoraria from Amgen and MEDPACE.
References
- 1) Ogura M, Okazaki S, Okazaki H, Tada H, Dobashi K, Nakamura K, Matsunaga K, Miida T, Minamino T, Yokoyama S, and Harada-Shiba M. Transition medicine in primary dyslipidemia. https://nanbyo-lipid.com/ (Japanese language)
- 2) Japan Pediatric Society. Recommendations for Transitional Care of Patients with Childhood-Onset Disease. J Jpn Pediatr Soc, 2014; 118; 98-106 (Japanese language)
- 3) Japan Pediatric Society. Recommendations to Promote Support for Transition to Adulthood for Patients with Childhood-Onset Chronic Diseases. J Jpn Pediatr Soc, 2023; 127; 61-78 (Japanese language)
- 4) Tada H, Kurashina T, Ogura M, Takegami M, Miyamoto Y, Arai H, Harada-Shiba M, Ishibashi S. Prospective Registry Study of Primary Dyslipidemia (PROLIPID): Rationale and Study Design. J Atheroscler Thromb, 2022; 29: 953-969
- 5) Available from the Website of the Information Center for Specific Pediatric Chronic Diseases, Japan. https://www.shouman.jp/ (Japanese language)
- 6) Tada H, Nomura A, Ogura M, Ikewaki K, Ishigaki Y, Inagaki K, Tsukamoto K, Dobashi K, Nakamura K, Hori M, Matsuki K, Yakashita S, Yokoyama S, Kawashiri MA, Harada-Shiba M. Diagnosis and Management of Sitosterolemia 2021. J Atheroscler Thromb, 2021; 28: 791-801
- 7) Kawashiri MA, Tada H. Sitosterolemia. Available from: https://nanbyo-lipid.com/wp/wp-content/themes/nanbyo_lipid/pdf/disease03_02.pdf (Japanese language)
- 8) Miyashita K, Lutz J, Hudgins LC, Toib D, Ashraf AP, Song W, Muramami M, Nakajima K, Ploug M, Fong LG, Young SG, Beigneux AP. Chylomicronemia from GPIHBP1 autoantibodies. J Lipid Res, 2020; 61: 1365-1376
- 9) Gaudet D, de Wal J, Tremblay K, Déry S, van Deventer S, Freidig A, Brisson D, Méthot J. Review of the clinical development of alipogene tiparvovec gene therapy for lipoprotein lipase deficiency. Atheroscler Suppl, 2010; 11: 55-60
- 10) Okazaki H, Gotoda T, Ogura M, Ishibashi S, Inagaki K, Daida H, Hayashi T, Hori M, Masuda D, Matsuki K, Yokoyama S, Harada-Shiba M; Committee on Primary Dyslipidemia under the Research Program on Rare and Intractable Disease of the Ministry of Health, Labour and Welfare of Japan. Current Diagnosis and Management of Primary Chylomicronemia. J Atheroscler Thromb, 2021; 28: 883-904
- 11) Gotoda T, Okazaki H. Primary Hyperchylomicronemia. Available from: https://nanbyo-lipid.com/wp/wp-content/themes/nanbyo_lipid/pdf/disease05_02.pdf (Japanese language)
- 12) Burnett JR, Hooper AJ, Hegele RA. Familial Lipoprotein Lipase Deficiency. GeneReviews® [Internet]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1308/
- 13) Brunzell JD, Deeb SS. Familial lipoprotein lipase deficiency, apoC-II deficiency, and hepatic lipase deficiency. The metabolic and molecular bases of inherited disease, 2001; 2: 2789-2816
- 14) Gotoda T, Shirai K, Ohta T, Kobayashi J, Yokoyama S, Oikawa S, Bujo H, Ishibashi S, Arai H, Yamashita S, Harada-Shiba M, Eto M, Hayashi T, Sone H, Suzuki H, Yamada N; Research Committee for Primary Hyperlipidemia, Research on Measures against Intractable Diseases by the Ministry of Health, Labour and Welfare in Japan. Diagnosis and management of type I and type V hyperlipoproteinemia. J Atheroscler Thromb, 2012; 19: 1-12
- 15) Williams L, Rhodes KS, Karmally W, Welstead LA, Alexander L, Sutton L; patients and families living with FCS. Familial chylomicronemia syndrome: Bringing to life dietary recommendations throughout the life span. J Clin Lipidol, 2018; 12: 908-919
- 16) Williams L, Wilson DP. Editorial commentary: Dietary management of familial chylomicronemia syndrome. J Clin Lipidol, 2016; 10: 462-465
- 17) Amundsen AL, Khoury J, Iversen PO, Bergei C, Ose L, Tonstad S, Retterstøl K. Marked changes in plasma lipids and lipoproteins during pregnancy in women with familial hypercholesterolemia. Atherosclerosis, 2006; 189: 451-457
- 18) Sekijima Y, Koyama S, Yoshinaga T, Koinuma M, Inaba Y. Nationwide survey on cerebrotendinous xanthomatosis in Japan. J Hum Genet, 2018; 63: 271-280
- 19) Appadurai V, DeBarber A, Chiang PW, Patel SB, Steiner RD, Tyler C, Bonnen PE. Apparent underdiagnosis of Cerebrotendinous Xanthomatosis revealed by analysis of ~60,000 human exomes. Mol Genet Metab, 2015; 116: 298-304
- 20) Koyama S, Sekijima Y, Ogura M, Hori M, Matsuki K, Miida T, Harada-Shiba M. Cerebrotendinous Xanthomatosis: Molecular Pathogenesis, Clinical Spectrum, Diagnosis, and Disease-Modifying Treatments. J Atheroscler Thromb, 2021; 28: 905-925
- 21) DeBarber AE, Duell PB. Update on cerebrotendinous xanthomatosis. Curr Opin Lipidol, 2021; 32: 123-131
- 22) Mignarri A, Gallus GN, Dotti MT, Federico A. A suspicion index for early diagnosis and treatment of cerebrotendinous xanthomatosis. J Inherit Metab Dis, 2014; 37: 421-429
- 23) Fiorucci S, Distrutti E. Chenodeoxycholic Acid: An Update on Its Therapeutic Applications. Handb Exp Pharmacol, 2019; 256: 265-282
- 24) Sekijima Y, Koyama S. Cerebrotendinous xanthomatosis. Available from: https://nanbyo-lipid.com/wp/wp-content/themes/nanbyo_lipid/pdf/disease06_02.pdf (Japanese language)
- 25) Burnett JR, Hooper AJ, Hegele RA. Abetalipoproteinemia. GeneReviews® [Internet]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK532447/
- 26) Japan Atherosclerosis Society. Diagnosis and Management of Hypolipidemia. 2017; Available from: https://www.j-athero.org/jp/wp-content/uploads/publications/pdf/shishitsuijou_2013_3_14.pdf (Japanese language)
- 27) Okazaki H, Takahashi M. Abetalipoproteinemia. Available from: https://nanbyo-lipid.com/wp/wp-content/themes/nanbyo_lipid/pdf/disease07_02.pdf (Japanese language)
- 28) Wakabayashi T, Takahashi M, Okazaki H. Familial Hypobetalipoproteinemia (FHBL) 1 (Homozygote). Available from: https://nanbyo-lipid.com/wp/wp-content/themes/nanbyo_lipid/pdf/ disease07_02.pdf (Japanese language)
- 29) Burnett JR, Hooper AJ, Hegele RA. APOB-Related Familial Hypobetalipoproteinemia. GeneReviews® [Internet]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK570370/
- 30) Bredefeld C, Hussain MM, Averna M, Black DD, Brin MF, Burnett JR, Charrière S, Cuerq C, Davidson NO, Deckelbaum RJ, Goldberg IJ, Granot E, Hegele RA, Ishibashi S, Karmally W, Levy E, Moulin P, Okazaki H, Poinsot P, Rader DJ, Takahashi M, Tarugi P, Traber MG, Filippo MD, Peretti N. Guidance for the diagnosis and treatment of hypolipidemia disorders. Journal of Clinical Lipidology, 2022; 16: 797-812
- 31) Kane JP, Havel RJ. Disorders of the Biogenesis and Secretion of Lipoproteins Containing the B Apolipoproteins. In: The Metabolic and Molecular Bases of Inherited Disease. 8th edition. Vol. 2. New York, NY: McGraw-Hill; 2001. 2717-2752
- 32) Berriot-Varoqueaux N, Aggerbeck LP, Samson-Bouma M, Wetterau JR. The role of the microsomal triglyceride transfer protein in abetalipoproteinemia. Annu Rev Nutr, 2000; 20: 663-697
- 33) Takahashi M, Okazaki H, Ohashi K, Ogura M, Ishibashi S, Okazaki S, Hirayama S, Hori M, Matsuki K, Yokoyama S, Harada-Shiba M. Current Diagnosis and Management of Abetalipoproteinemia. J Atheroscler Thromb, 2021; 28: 1009-1019
- 34) Burnett JR, Bell DA, Hooper AJ, Hegele RA. Clinical utility gene card for: Familial hypobetalipoproteinaemia (APOB)--Update 2014. Eur J Hum Genet, 2015; 23(6)
- 35) Harada-Shiba M, Ohtake A, Sugiyama D, Tada H, Dobashi K, Matsuki K, Minamino T, Yamashita S, Yamamoto Y. Guidelines for the Diagnosis and Treatment of Pediatric Familial Hypercholesterolemia 2022. J Atheroscler Thromb, 2023; 30: 531-557
- 36) Tada H, Hori M, Nomura A, Hosomichi K, Nohara A, Kawashiri MA, Harada-Shiba M. A catalog of the pathogenic mutations of LDL receptor gene in Japanese familial hypercholesterolemia. J Clin Lipidol, 2020; 14: 346-351 e9
- 37) Hori M, Takahashi A, Son C, Ogura M, Harada-Shiba M. The first Japanese cases of familial hypercholesterolemia due to a known pathogenic APOB gene variant, c.10580 G>A: p.(Arg3527Gln). J Clin Lipidol, 2020; 14: 482-486
- 38) Familial Hypercholesterolemia (No. 130). Information Center of the Specific Pediatric Chronic Diseases. Available from: https://www.shouman.jp/disease/details/08_12_130/ (Japanese language)
- 39) Familial Hypercholesterolemia (Homozygote) (No. 79). Information Center of the Intractable Diseases. Available from: https://www.nanbyou.or.jp/entry/226 (Japanese language)
- 40) Harada-Shiba M, Arai H, Ohmura H, Okazaki H, Sugiyama D, Tada H, Dobashi K, Matsuki K, Minamino T, Yamashita S, Yokote K. Guidelines for the Diagnosis and Treatment of Adult Familial Hypercholesterolemia 2022. J Atheroscler Thromb, 2023; 30: 558-586
- 41) Harada-Shiba M, Ohta T, Ohtake A, Ogura M, Dobashi K, Nohara A, Yamashita S, Yokote K; Joint Working Group by Japan Pediatric Society and Japan Atherosclerosis Society for Making Guidance of Pediatric Familial Hypercholesterolemia. Guidance for Pediatric Familial Hypercholesterolemia 2017. J Atheroscler Thromb, 2018; 25: 539-553
- 42) Mehta R, Elias-Lopez D, Martagon AJ, Pérez-Méndez OA, Ordóñez Sánchez ML, Segura Y, Tusié MT, Aguilar-Salinas CA. LCAT deficiency: a systematic review with the clinical and genetic description of Mexican kindred. Lipids Health Dis, 2021; 20: 70
- 43) Kuroda M, Bujo H, Yokote K, Murano T, Yamaguchi T, Ogura M, Ikewaki K, Koseki M, Takeuchi Y, Nakatsuka A, Hori M, Matsuki K, Miida T, Yokoyama S, Wada J, Harada-Shiba M. Current Status of Familial LCAT Deficiency in Japan. J Atheroscler Thromb, 2021; 28: 679-691
- 44) Vitali C, Bajaj A, Nguyen C, Schnall J, Chen J, Stylianou K, Rader DJ, Cuchel M. A systematic review of the natural history and biomarkers of primary lecithin: cholesterol acyltransferase deficiency. J Lipid Res, 2022; 63: 100169
- 45) Pavanello C, Calabresi L. Genetic, biochemical, and clinical features of LCAT deficiency: update for 2020. Curr Opin Lipidol, 2020; 31: 232-237
- 46) Miller WG, Myers GL, Sakurabayashi I, Bachmann LM, Caudill SP, Dziekonski A, Edwards S, Kimberly MM, Korzun WJ, Leary ET, Nakajima K, Nakamura M, Nilsson G, Shamburek RD, Vetrovec GW, Warnick GR, and Remaley AT. Seven Direct Methods for Measuring HDL and LDL Cholesterol Compared with Ultracentrifugation Reference Measurement Procedures. Clin Chem, 2010; 56: 977-986
- 47) Bujo H, Kuroda M, Murano T. Lecithin cholesterol acyltransferase deficiency. Available from: https://nanbyo-lipid.com/wp/wp-content/themes/nanbyo_lipid/pdf/disease02_02.pdf (Japanese language)
- 48) Naito S, Kamata M, Furuya M, Hayashi M, Kuroda M, Bujo H, Kamata K. Amelioration of circulating lipoprotein profile and proteinuria in a patient with LCAT deficiency due to a novel mutation (Cys74Tyr) in the lid region of LCAT under a fat-restricted diet and ARB treatment. Atherosclerosis, 2013; 228: 193-197
- 49) Yee MS, Pavitt DV, Richmond W, Cook HT, McLean AG, Valabhji J, Elkeles RS. Changes in lipoprotein profile and urinary albumin excretion in familial LCAT deficiency with lipid-lowering therapy. Atherosclerosis, 2009; 205: 528-532
- 50) Gjone E. Familial lecithin: cholesterol acyltransferase deficiency--a clinical survey. Scand J Clin Lab Invest Suppl, 1974; 137: 73-82
- 51) Koseki M, Yamashita S, Ogura M, Ishigaki Y, Ono K, Tsukamoto K, Hori M, Matsuki K, Yokoyama S, Harada-Shiba M. Current Diagnosis and Management of Tangier Disease. J Atheroscler Thromb, 2021; 28: 802-810
- 52) Koseki M, Yamashita S. Tangier disease. Available from: https://nanbyo-lipid.com/wp/wp-content/themes/nanbyo_lipid/pdf/disease04_02.pdf (Japanese language)
- 53) Schaefer EJ, Anthanont P, Diffenderfer MR, Polisecki E, Asztalos BF. Diagnosis and treatment of high-density lipoprotein deficiency. Prog Cardiovasc Dis, 2016: 97-106
- 54) Eto M, Saito M, Nakata H, Iwashima Y, Watanabe K, Ikoda A, Kaku K. Type III hyperlipoproteinemia with apolipoprotein E2/2 genotype in Japan. Clin Genet, 2002; 61: 416-422
- 55) Ohta T, Kiwaki K, Endo F, Umehashi H, Matsuda I. Dyslipidemia in young Japanese children: its relation to familial hypercholesterolemia and familial combined hyperlipidemia. Pediatrics International, 2002; 44: 602-607
- 56) Iwata F, Okada T, Kuromori Y, Hara M, Harada K. Screening for familial combined hyperlipidemia in children using lipid phenotypes. J Atheroscler Thromb, 2003; 10: 299-303
- 57) Matsunaga K, Mizobuchi A, Fu HY, Ishikawa S, Tada, H, Kawashiri MA, Yokota I, Sasaki T, Ito S, Kunikata J, Iwase T, Hirao T, Yokoyama K, Hoshikawa Y, Fujisawa T, Dobashi K, Kusaka T and Minamino T. Universal Screening for Familial Hypercholesterolemia in Children in Kagawa, Japan. J Atheroscler Thromb, 2022; 29: 839-849
- 58) Takeji Y, Tada H, Ogura M, Nohara A, Kawashiri MA, Yamashita Y, Harada-Shiba M, and on behalf of the Committee on Primary Dyslipidemia under the Research Program on Rare and Intractable Disease of the Ministry of Health, Labour and Welfare of Japan. Clinical Characteristics of Homozygous Familial Hypercholesterolemia in Japan. A Survey Using a National Database, JACC: Asia. 2023; 3: 881-891
- 59) Luilink IK, Wiegman A, Kusters DM, Hof MH, Groothoff JW, de Groot E, Kastelein JJP, Hutten BA. 20-Year Follow-up of Statins in Children with Familial Hypercholesterolemia. N Engl J Med, 2019; 381: 1547-1556

