Review
High-altitude Pulmonary Edema: Review
2014 年 56 巻 4 号 p. 235-243

詳細
2014 年 56 巻 4 号 p. 235-243
Objective: At High altitude (HA) (elevation >2,500 m), hypobaric hypoxia may lead to the development of symptoms associated with low oxygen pressure in many sojourners. High-altitude pulmonary edema (HAPE) is a potentially fatal condition, occurring at altitudes greater than 3,000 m and affecting rapidly ascending, non-acclimatized healthy individuals. It is a multifactorial disease involving both environmental and genetic risk factors. Since thousands of lowlanders travel to high altitude areas for various reasons every year, we thought it would be interesting to review pathological aspects related to hypobaric hypoxia, particularly HAPE. Method: Since the pathogenesis of HAPE is still a subject of study, we systematically identified and categorized a broad range of facets of HAPE such as its incidence, symptoms, physiological effects, pathophysiology including physiological and genetic factors, prevention and treatment. Results: This review focuses on HA-related health problems in general with special reference to HAPE, which is one of the primary causes of deaths at extreme altitudes. Hence, it is extremely important, as it summarizes the literature in this area and provides an overview of this severe HA malady for evaluation of physiological, biochemical and genetic responses during early induction and acclimatization to HA. This article could be of broad scientific interest for researchers working in the field of high altitude medicine.
(J Occup Health 2014; 56: 235-243)
Mountains cover over one-fifth of the earth's surface and are popular tourist destinations. With the advent of better transportation, access to moderate (2,000 to 4,000 m) and even very high altitudes (>5,500 m) has become increasingly convenient. High-altitude regions are home to nearly three hundred million people in the world, of which around 140 million people live above 2,400 m. The Andes and the highlands of Ethiopia (which are home to approximately 12 million people) are more densely populated (approximately 50 million people), and the four Chinese plateaus are home for nearly 60–80 million people. Large numbers of people travel to high altitudes for various reasons, such as trekking, mining, pilgrimage, tourism and deployment, as in the case of military personnel deployed to high altitudes to guard the borders of a country. Exposure to the oxygen-depleted environment triggers the onset of a range of physiological and biochemical reactions that primarily gear the individual towards enhancement of the efficacy of the body's respiratory1), cardiovascular and oxygen utilization systems2) through the process of acclimatization.
Altitude acclimatization can have immediate effects such as hyperventilation, fluid loss due to a decreased thirst drive, increased heart rate and slightly lowered stroke volume, whereas long-term effects include compensatory alkali loss in urine, increased hematocrit (polycythemia), higher concentration of capillaries in skeletal muscle tissue, increased myoglobin and hypoxic pulmonary vasoconstriction.
“Altitude sickness” is a commonly used term for syndromes encountered at an altitude above 2,500 m, comprising acute mountain sickness (AMS), high-altitude cerebral edema (HACE) and high-altitude pulmonary edema (HAPE). Rate of ascent, altitude reached, pre-acclimatization and individual susceptibility are the major determinants of susceptibility to high-altitude maladies. Besides these, other high-altitude diseases include chronic mountain sickness, subacute mountain sickness and the less severe retinal hemorrhage. Studies on different populations have suggested variability in individual susceptibility to altitude sickness depending on genetic makeup3).
HAPE is a potentially fatal medical condition and which is the cause of most of the deaths due to high-altitude illnesses4). It is a form of accelerated, permeability pulmonary edema of non-cardiogenic origin usually occurring within 2–4 days of ascent above 2,500–3,000 m4–6); it rarely occurs after more than 4 or 5 days at the same altitude, most likely because of remodeling and adaptation7). HAPE is associated with pulmonary hypertension and elevated capillary pressure8). It can manifest in two forms: it can occur in inhabitants returning from a sojourn at a low altitude, also known as “re-entry” HAPE, and it can occur in rapidly climbing unacclimatized healthy lowlanders. It is considered as a multifactorial condition whose inception and progression are governed both by genetic and environmental factors3). The first modern English language report of HAPE, by Houston9) was in 1960. Individuals, who ascend rapidly above 4,500 m with a previous history of HAPE have a 60% chance of HAPE recurrence10). It has been reported that a previous history of HAPE can predict its recurrence11).
The occurrence of HAPE at high altitudes is reported from various parts of the world. A study conducted on 52 patients admitted to a hospital between 1992 and 2000 in the French Alps reported HAPE at moderate altitudes of between 1,400–2,400 m12), though the incidence of HAPE at these altitudes is considered rare. One of the earliest reports of re-entry HAPE came from Peru, which described it as a pulmonary edema associated with electrographic signs of right ventricular overload13).
Owing to the interest of sojourners, individuals deployed for defense in high altitude regions, pilgrimages, etc. and due to the criticality of this high-altitude malady, the aim of this review article was to summarize the current literature and present the relevant details to researchers interested in HAPE and readers in general.
Incidence: In 1970s, the incidences of HAPE were reported to be 0–6% in adults and 8–9% in children in Peru14). Sophocles (1986), in his study of HAPE cases from 1975 to 1982, estimated the incidence of HAPE in visitors to ski resorts in Vail, Colorado which is in the Rocky Mountains, (elevation 2,500 m) to be 0.01–0.1%15). His study also reported that men are more susceptible to HAPE as compared with women15). A lower incidence of HAPE in women was also reported by Hultgren et al. (1996) in their study on HAPE patients in the Rocky Mountains in Colorado, where 84% of the HAPE patients were men as compared with women16). In the same study, the average arterial oxygen saturation in patients with HAPE was 74% as compared with a normal average oxygen saturation of 92% at such altitude, showing the occurrence of hypoxemia in these patients.
Symptoms: The symptoms of HAPE may appear over the course of several hours or days; however, sometimes the symptoms appear suddenly after a night's sleep at a high altitude. Although HAPE can occur without preceding AMS17), HAPE is commonly found to develop in association with AMS18). Early symptoms of HAPE include shortness of breath, exertional dyspnoea, fatigue, cough, headache, tightness of the chest or congestion, a sudden reduction in exercise performance and elevated body temperature, generally not exceeding 38.5°C. As pulmonary edema progresses, cough becomes aggravated and breathlessness is felt even at rest. Gurgling sounds from the chest and pink frothy sputum along with cyanosis indicates advanced cases. Hypoxemia and respiratory alkalosis are revealed by arterial blood gas measurements4). Signs include less or no sputum and non-specific findings on chest X-ray19). In advanced cases, HAPE may be associated with HACE (characterized by swelling of brain tissue due to fluid leakage) and show symptoms of cerebral edema (such as ataxia and decreased levels of consciousness.
The most popular of the proposed mechanisms of high-altitude pulmonary edema includes uneven hypoxic pulmonary vasoconstriction that exposes pulmonary capillaries to a high pressure, damaging their walls and leading to a high-permeability form of edema. Studies have shown that the hypoxic environment at high altitudes impairs physical performance, sleep and mental performance20); however, these problems do not have a proven pathological background.
Maximal consumption of oxygen: The reduced maximal oxygen consumption at high altitudes is usually due to the reduction in mitochondrial pO2, which inhibits the function of the electron transport chain responsible for providing cellular energy during oxidative phosphorylation, though the exact pathway still remains to be elucidated (Fig. 1). At an altitude of 3,000 m, maximal oxygen consumption is reduced to approximately 85%21). Due to this, fatigue occurs, leading to reduced physical performance22). It has also been demonstrated that individuals with the insertion allele rather than the deletion variant in the angiotensin-converting enzyme (ACE) gene tend to have better maximal oxygen uptake at high altitudes23).
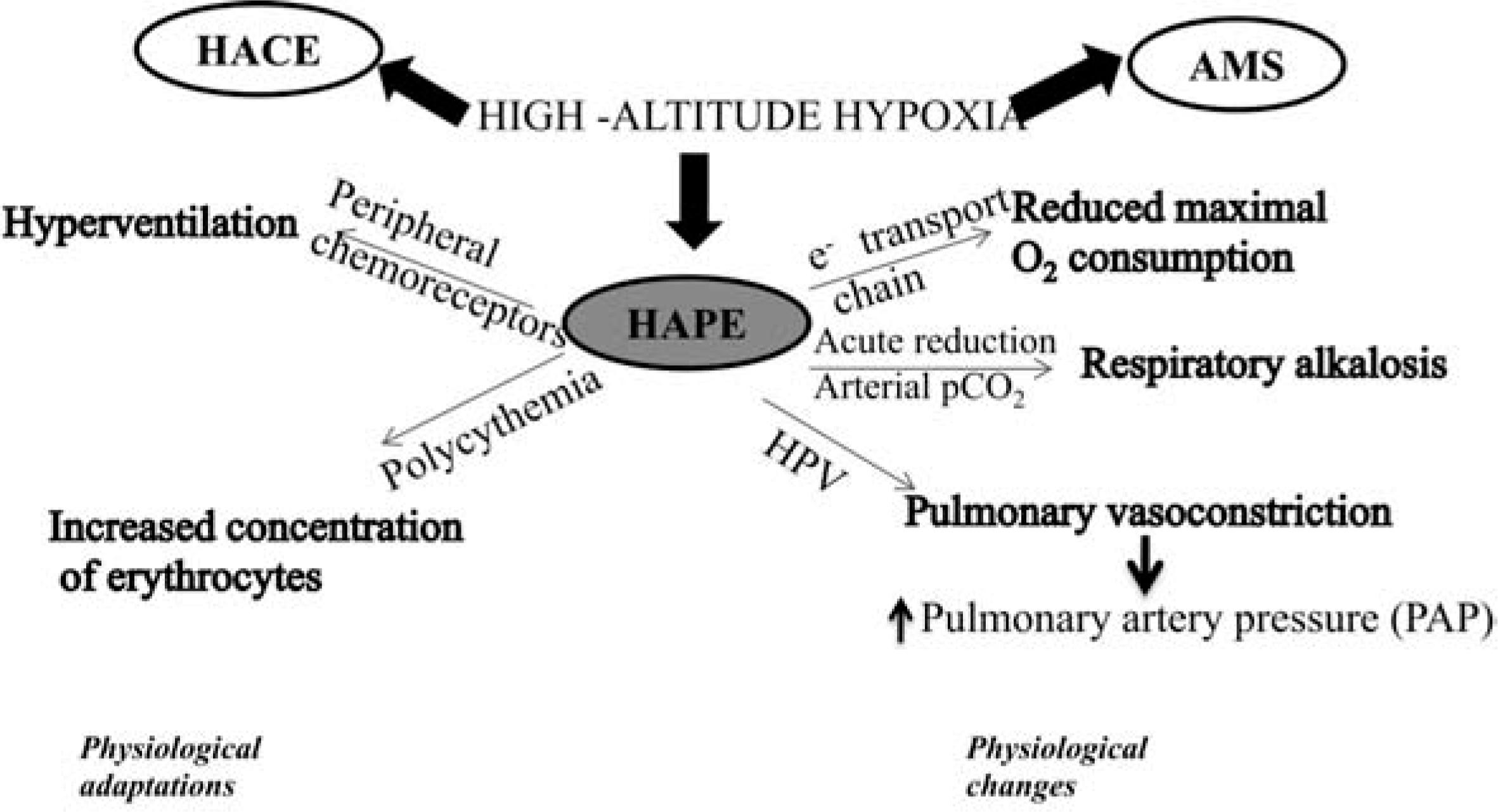
Summary of physiological changes and adaptations as a result of high-altitude exposure.
Polycythemia: This refers to an increased concentration of erythrocytes, which helps to increase the oxygen carrying capacity of blood amongst lowlanders (natives of sea level) staying at high altitudes for longer durations and highlanders (natives of high altitudes) except for Tibetans and related groups (like Sherpas). This process may take several weeks to complete; it develops relatively slow and may take several days before an increased rate of erythrocyte production is evident24). Therefore, polycythemia does not seem to play an important role in rapid acclimatization to high altitudes20) (Fig. 1). Low temperatures at high altitudes lead to decreased thirst drive, which in turn leads to dehydration and a reduced plasma concentration. This reduced plasma volume results in an increase in the concentration of erythrocytes per unit blood volume.
Hyperventilation: This term refers to an increase in the rate and depth of breathing at high altitudes resulting in increased alveolar ventilation. Hypoxic environments result in hyperventilation and a reduction in pCO2, as recognized by Rahn and Otis in 194925). The physiological advantage of hyperventilation is that it lessens the otherwise occurring fall in alveolar pO2. The extent of hyperventilation at high altitudes can be quiet noticeable, e.g., on the summit of Mount Everest, where the inspired pO2 is only 29% of its sea level value, alveolar ventilation is increased by ∼5 fold. This results in reduction of the pCO2 to 7 to 8 mm Hg, one-fifth of its normal sea level value of 40 mm Hg, and maintenance of the alveolar pO2 at nearly 35 mm Hg, which is low but sufficient to keep the climber alive26).
Acid-base changes: An acute reduction in alveolar arterial pCO2 causes respiratory alkalosis with an increased pH in both the cerebrospinal fluid and arterial blood (Fig. 1). The initial alkalosis occurs in both; cerebrospinal fluid and blood, which tends to inhibit hyperventilation. Oxygen sensors in the carotid body initiate a hypoxic ventilator response27) that helps to compensate for the oxygen deficit despite the risk of alkalosis. After one or two days, the pH of the cerebrospinal fluid changes towards normal as a result of movement of bicarbonate ions out of cerebrospinal fluid, and after two to three days, the pH of the arterial blood moves towards normal as a result of renal excretion of bicarbonate1).
Pathophysiology of HAPESeveral hypotheses have been suggested to explain the mechanism underlying the origin of HAPE, although its exact cause remains unknown. Its pathophysiology is considered to be initiated by the increased pressure in the pulmonary capillaries during prolonged altitude exposure8, 28). This pressure is the force driving fluid out of the pulmonary capillaries due to interstitium leakage, which can be a contributing factor to edema formation29).
The increasing pressure on the capillaries imparts excessive stress on the collagen and the extracellular matrix of the alveolar capillary barrier, which in turn leads to the mechanically induced breaks in the blood gas barrier, also described as the “stress failure” of pulmonary capillaries by West and colleagues (1991)30). A study using a rat model by Bai et al. in 2010 showed a direct relation between the histological evidence of pulmonary capillary stress failure and progression of an illness similar to HAPE31). Rats exposed to physiologically relevant levels of hypobaric hypoxia developed elevated bronchoalveolar lavage (BAL) fluid total protein, albumin and red blood cell concentrations. These changes were more evident in animals subjected to higher levels of physical exercise under hypoxic conditions and were not observed in normoxic resting animals31).
The classical concept of a characteristic increase in hypoxic pulmonary vasoconstriction (HPV) is also proposed as an important pathogenic factor in the development of HAPE32, 33). Alveolar hypoxia leads to an adaptive vasomotor response in the form of HPV, which is non-homogenous in nature. The pulmonary capillary pressure increases as a result of HPV, which occurs mainly in smaller pulmonary arteries34). As a result of the constriction of small pulmonary arteries, blood gets diverted away, causing elevated blood flow and raising the pressure, which consequently leads an the increase in capillary permeability. Endothelial dysfunction and sympathetic activation as observed in animals and human models are considered the prime contributors to exaggerated HPV32, 35). It has been observed that most HAPE-susceptible individuals have a lowered hypoxic ventilator response, which leads to a low alveolar pO22, 36). The regional over-perfusion in the lungs resulting from non-homogenous HPV37) contributes to the development of HAPE, mainly in areas of elevated pressure. The peripheral chemoreceptors located on the pulmonary vasculature detect the drop in alveolar oxygen tension and lead to vasoconstriction of smaller pulmonary arteries38, 39) and pulmonary veins40). The smooth muscle cells of the pulmonary vasculature respond to acute hypoxia within seconds. This involves the inhibition of voltage-dependent potassium channels, causing membrane depolarization and calcium entry through voltage-gated L-type calcium channels39, 41).
Individuals developing HAPE have an abnormal rise in pulmonary artery pressure (PAP) during brief or prolonged exposure to hypoxia. A reduced availability of nitric oxide has also been suggested to be a major cause of elevated PAP28). Increased heterogeneity in pulmonary blood flow has been demonstrated in HAPE-susceptible individuals who had HAPE at least once in their lifetime as compared with non-susceptible controls who had not developed HAPE even when repeated exposure to altitudes greater than 5,400 m42).
Under the conditions of hypoxia, expression of the EPO gene (erythropoietin) is activated via binding of hypoxia inducible factor-1 (HIF-1). Synthesis of erythropoietin gene is regulated at both transcriptional and post-transcriptional levels. Increased secretion of erythropoietin in response to hypoxia has been reported in HAPE patients, which could be due to exaggerated hypoxemia in these patients43).
Mechanism of edema formation in HAPE: The location of fluid leakage caused by hypoxia-induced pulmonary vasoconstriction is not known. Pulmonary vasoconstriction during HAPE is heterogenous and unevenly distributed. It is evident by asymmetric distribution of rales and infiltrates, as seen during clinical examinations performed early in the course of edema. In animals exposed to a hypoxic environment, small arterioles are the site of transvascular leakage as a result of increased PAP44). Pulmonary veins also contract in response to hypoxia45), thus increasing the resistance in the fluid filtration region. But the vessels that are not constricted may be subjected to high-pressure flow and are relatively over-perfused as compared with other vessels46).
Alveolar fluid clearance: The weakened capacity of alveolar fluid reabsorption is also considered a mechanism contributing to the pathophysiology of HAPE. The concept of alveolar flooding associated with increased pulmonary hypertension, as a constituting factor of HAPE, has been shown to be related to a defect in respiratory sodium and water transport. Due to the inability to assess alveolar transport activity directly, nasal transepithelial Na+ transport activity is used as a surrogate marker to study the ion transport. Hypoxic conditions of high altitudes hamper nasal transepithelial Na+ transport, thus impairing fluid absorption in the human lungs47). Decreased potential difference and reduced Na+ transport were reported in HAPE-susceptible subjects as predisposing factor to HAPE in the in vivo study done by Sartori et al. They demonstrated that an environmental factor may impair respiratory transepithelial sodium transport in humans47), thus suggesting that a combination of genetic and environmental mechanisms facilitates HAPE. Keeping in view the possible role of reduced alveolar fluid clearance in HAPE, prophylactic inhalation of the adrenergic agonist salmeterol, which up-regulates the clearance of alveolar fluid in animal models, was studied, and it was shown to reduce the incidence of HAPE by 50%47, 48).
Nature of fluid leakage: Swenson et al (2002), in their study using BAL performed within a day of ascent to 4,559 m, showed that early HAPE is characterized by a protein-rich fluid containing albumin in high concentrations and an elevated red blood cell count, both in clinically ill HAPE subjects and subjects who developed HAPE in the next 24 hours48). They demonstrated that this leakage in early HAPE is noninflammatory and a unidirectional breach of the alveolar capillary barrier in the absence of inflammation. Pulmonary edema in HAPE was thus concluded to be hydrostatic with altered alveolar-capillary permeability49). At a later stage, increased alveolar macrophages, neutrophils and concentrations of inflammatory mediators (IL-1, TNF alpha, IL-8, prostaglandin E2, etc.) occur.
Amongst lowlanders ascending to high altitudes, genetic differences have been identified that confer performance benefits at high altitudes. Exploring these differences by studying humans exposed to high-altitude hypoxia has the potential to identify mechanisms important in established critical illnesses and perhaps to alter our therapeutic focus towards increasing the efficiency of oxygen utilization rather than improving delivery50).
The response of cells to low oxygen (hypoxia) is characterized by coordinated regulation of the expression of a large number of genes with widespread functions such as energy metabolism, vascular supply and growth51). Although genetic variation provides a possible mechanism to explain inter-individual differences in performance in response to hypoxia/susceptibility to HAPE and a significant association has been identified in the polymorphic variants of these genes and occurrence/sensitivity to HAPE, however, the definite role of these SNPs has yet to be established. Some recent studies have shown a possible association between the polymorphisms existing in the genes of the RAAS pathway (rennin-angiotensin-aldosterone system) playing a key role in regulation of vascular tone circulatory homeostasis) and sensitivity of an individual to develop HAPE52, 53).
Hypoxia-inducible factor-1 (HIF-1) is a basic transcription factor that is involved in many important homeostatic responses to hypoxia. Genes that are transcriptionally activated by HIF-1 include; erythropoietin (EPO), a regulator of erythropoiesis and a major determinant of oxygen carrying capacity of blood54); genes encoding glycolytic enzymes such as aldolase A (ALDA), enolase I (ENO1), lactate dehydrogenase A (LDHA) and phosphofructokinase L (PFKL), which provide a metabolic pathway for ATP generation in absence of O255, 56); heme oxygenase 1 (HO1) and inducible nitric oxide synthase (iNOS) which are responsible for synthesis of vasoactive molecules carbon monoxide and nitric oxide; and vascular endothelial growth factor (VEGF), an important regulator of angiogenesis and thus a major determinant of tissue perfusion57) (Fig. 2).
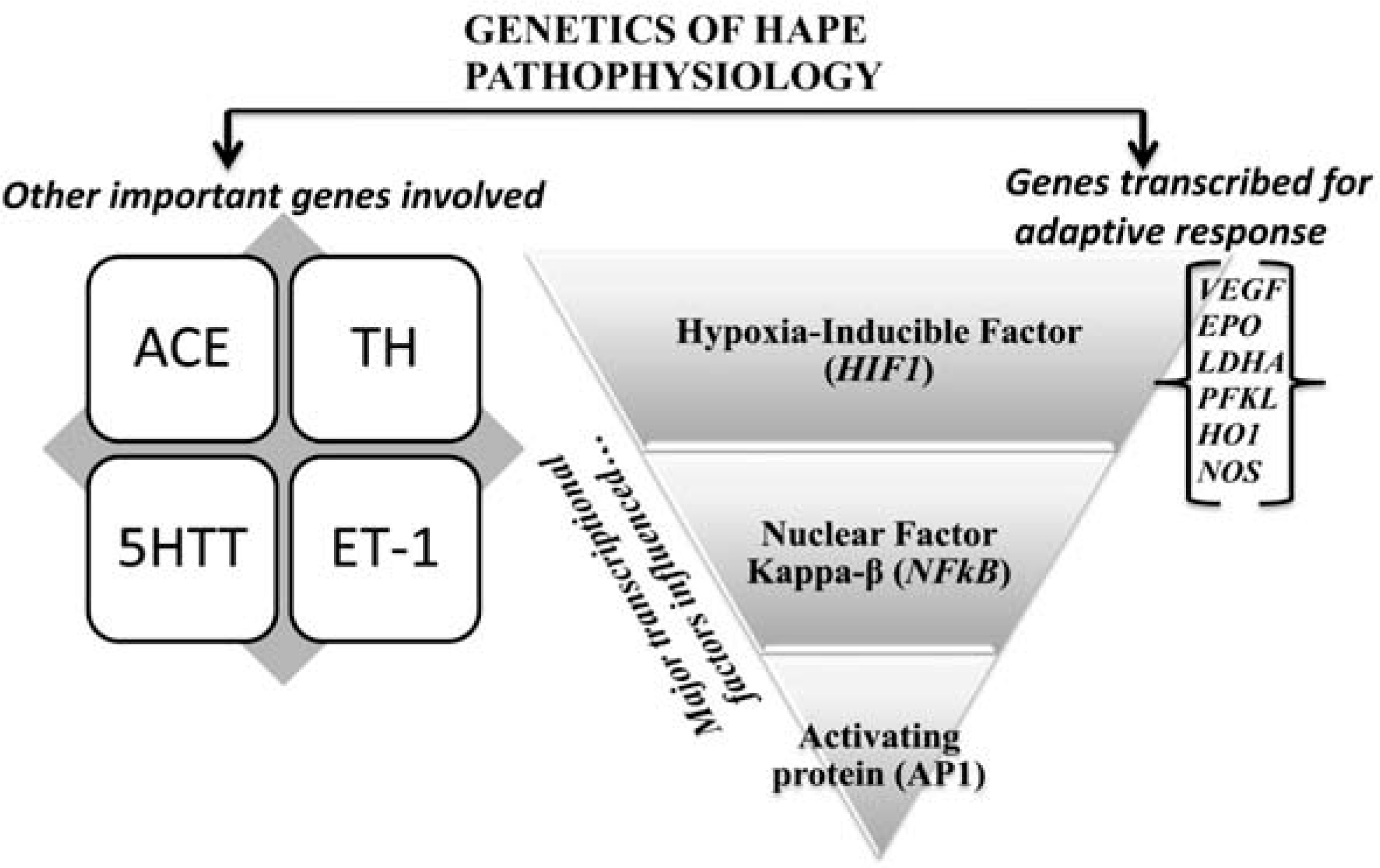
Genetic factors involved in high-altitude hypoxic adaptation/maladaptation.
Also, genes such as angiotensin-1 converting enzyme (ACE); tyrosine hydroxylase (TH) (the key enzyme that catalyses the hydroxylation of tyrosine to form dihydroxyphenylalanine (dopa)), serotonin transporter (5-HTT) (a potent bronchoconstrictor58), a mitogen for pulmonary smooth muscle cells59) and a vasodilator in the systemic circulation60)), endothelial nitric oxide synthase (eNOS) (a potent vasodilator); and endothelin-1 (ET-1) (a potent vasoconstrictor) seem to have a possible involvement in physiological adaptation to HAPE (Fig. 2).
Adaptation to hypoxia in cells, tissues and the organism as a whole depends largely on the changes in the expression of genes encoding a diverse group of physiologically relevant proteins. The functional involvement of these genes in acclimatization to hypoxic environment is detailed below.
Nitric oxide synthase (NOS): This gene exists in three forms, neuronal NOS (NOS I), inducible NOS (NOS II) and endothelial NOS (NOS III). NOS I and NOS III are constitutively expressed and produce NO for basal blood flow and pressure regulation across many organs and species. Droma et al. (2002) have shown significant associations of mutations occurring in the eNOS gene in conjunction with HAPE, and a genetic background may underlie the impaired NO synthesis in the pulmonary circulation of HAPE-sensitive patients. They concluded that these single-nucleotide polymorphisms in the eNOS gene could be genetic markers for predicting the susceptibility to HAPE61). eNOS gene polymorphisms have also been shown to be associated with susceptibility to HAPE in the Chinese population62). However, these studies do not give definite conclusive statements.
Tyrosine hydroxylase (TH): TH is the key enzyme that catalyses the hydroxylation of tyrosine to form dihydroxyphenylalanine, which is the first and rate-limiting step in the biosynthesis of catecholamine neurotransmitters63). The peripheral chemoreceptors for hypoxia are located in the carotid bodies (CBs)64). When CB glomus cells are stimulated by hypoxia, they release catecholaminergic neurotransmitter dopamine that subsequently emanates neuronal signals to a higher ventilatory center, resulting in hyperventilation. This role of TH indicates that the polymorphisms occurring in this gene might lead to a decreased peripheral chemoreceptor dopamine synthesis, which might blunt HVR and make an individual susceptible to HAPE. Most commonly, a microsatellite marker, a tetranucleotide repeat (TCAT)n, located in the first intron with at least five individual alleles has been identified65), along with an amino acid variant swapping valine (Val) for methionine (Met) at codon 81 in exon 2 of the TH gene66), playing vital role in its expression.
Angiotensin-converting enzyme (ACE): In the search for the inheritable determinants of HAPE, the gene encoding ACE ranks high. ACE catalyses generation of angiotensinogen II and has the ability to modulate the pulmonary vasoconstrictive response to hypoxia via the interaction with its receptor67). There is wide interindividual variability in the plasma ACE levels, and this variability might be explained by the insertion/deletion (I/D) polymorphism of the human ACE gene. Qi et al. (2011) conducted meta-analysis of consolidated data to study the association between the angiotensin-converting enzyme (ACE) I/D polymorphism and HAPE. Their results supported the notion that ACE D allele carriers were at significant increased risk of developing HAPE68). The I allele has been associated with lowered ACE protein levels compared with the D allele69). Woods et al. (2002) suggested that individuals with the I/I genotype probably maintain a higher arterial oxygen saturation (SaO2) at rest and also during exercise at high altitudes compared with carriers of the D allele, thereby providing an edge to individuals for better performance under extreme conditions found at high altitudes70).
Endothelin 1: The product of the endothelin-1 gene (EDN-1) is a 21-residue polypeptide that is synthesized and secreted by vascular endothelial cells. It is a potent endogenously produced vasoconstrictor. Combined actions of EDN-1 and nitric oxide might play an important role in regulating vascular tone and blood pressure. EDN-1 is considered a contributor to the pathogenesis of HAPE71, 72). The findings of Sartori et al. (1999) suggest that in HAPE-susceptible mountaineers, an augmented release of the potent pulmonary vasoconstrictor peptide endothelin-1 and/or its reduced pulmonary clearance could represent one of the mechanisms contributing to exaggerated pulmonary hypertension at high altitudes71). A study involving 426 highlanders and 236 lowlanders reported an over-representation of longer repeats, -3A/-3A, GG and Lys198Lys genotypes of EDN1 along with low plasma endothelin levels in the highlanders compared with the lowlanders, suggesting a crucial role in high-altitude acclimatization73). When these EDN1 variants along with ACE (I/D) were studied, higher ACE activity and endothelin levels were reported in HAPE patients.
Vascular endothelial growth factor: VEGF is an important regulator of angiogenesis having a number of consensus HIF-1 binding sites are present in the 5’-flanks of the VEGF gene. Findings of Hanaoka et al. (2003) suggest that VEGF is probably destroyed in the lungs of HAPE patients, and it does not appear to play a critical part in the pathogenesis of HAPE but has rather an important role in the repair process for the impaired cell layer74).
The most commonly studied VEGF polymorphism is C(936)T, and it has been reported that subjects carrying the VEGF 936T allele had significantly lower plasma VEGF levels as compared with subjects carrying the VEGF 936CC genotype75). In a study on the Japanese population, C(936)T polymorphism in the 3’UTR region along with C(−2578)A, G(−1154)A and T(−460)C in the promoter region have not been found to be associated with susceptibility to HAPE76).
Ascending slowly is the most effective way to prevent HAPE and is even useful in susceptible individuals. However, there have been no studies establishing the incidence of HAPE according to rate of ascent. Climbers with any symptoms of AMS or initiation of HAPE are advised not to ascend further and to avoid exercise during first few days of exposure, as exercise may enhance or cause pulmonary edema.
One of the prevention regimens is the use of the drug nifedipine (a calcium channel blocker and an inhibitor of hypoxic pulmonary vasoconstriction); it causes pulmonary vasodilation and as a result limits the rise in PAP that contributes to edema formation32). Sildenafil, which reduces hypoxic pulmonary vasoconstriction and has been studied for the pulmonary vascular response to hypoxia in humans and mice, where it attenuated hypoxia-induced pulmonary hypertension77), is also effective for prevention and treatment of HAPE. Dexamethasone also significantly attenuates the increase in PAP at high altitudes11). The suggested dose of dexamethasone is 8 mg initially followed by 4 mg every 6 hours. It is also useful for relief of cerebral symptoms of severe mountain sickness78). Prevention of an excessive rise in PAP is necessary for prevention of HAPE. Nitric oxide inhalation has been shown to improve arterial oxygenation and reduce the hypoxia-induced rise in PAP in HAPE subjects and to rapidly improved arterial oxygenation34). A combined use of inhaled nitric oxide (NO) and oxygen has been shown to have an additive effect on pulmonary hemodynamics and to facilitate gas exchange. Inhalation of nitric oxide redistributes blood flow in the lungs away from edematous regions toward non-edematous regions, thereby improving arterial oxygenation in HAPE-prone subjects20, 34).
The rational treatment for HAPE requires increase of alveolar PO2 by administration of supplementary oxygen to maintain arterial saturation above 90%, accompanied by rest from strenuous physical activity. Advanced cases of HAPE should be brought immediately to low altitudes for immediate relief. If urgent descent is not possible, portable hyperbaric bags such as the Gamow bag can be used for both high-altitude cerebral edema and high-altitude pulmonary edema20).
In this review, we have made an effort to detail the diversity of physiological and genetic responses to hypoxia among patients with high-altitude pulmonary edema and summarized various studies showing significant differences in the amplitude and breadth of hypoxic responses. The reports presented here depicts early responses on ascent to high altitudes, the pathophysiology of HAPE and the roles of candidate gene polymorphisms in the pathogenesis of HAPE along with its prevention and treatment, which would be helpful in better understanding this high-altitude malady and formulating strategies to deal with it.
Conflict of interest: The authors declare that they have no conflicts of interest.