Abstract
Diplonemea is one of the most abundant and species-rich protist groups in marine environments. However, many lineages are still undescribed. Particularly, little is understood about members of the ‘deep-sea and pelagic diplonemids (DSPD)’ clade. The environmental DNA (eDNA) studies conducted with universal eukaryotic primers have shown that DSPDs were also abundantly distributed in natural environments, but their cultures have not yet been established. For future studies such as mitochondrial genome sequencing, culture establishment is needed. For establishing cultures, it is important to select samples containing a high quantity of diplonemids. In this study, we designed new diplonemid primers for eDNA analysis using a next-generation sequencer, testing its efficiency using eDNA that was extracted from two deep-sea water samples. Out of a total of 58,154 assembled reads, 57,633 reads (i.e. 99.1%) were affiliated with diplonemids by BLAST and reconstructed into 160 representative sequences. Phylogenetic analyses showed that many of the representative sequences (137 sequences, 85.6%) were branched within the DSPD clade and family Hemistasiidae. These findings indicate that the new primers are useful in monitoring diplonemid diversity and acquiring information for the establishment of DSPD cultures.
INTRODUCTION
Diplonemea is a subgroup of Euglenozoa and composed of heterotrophic biflagellates (Adl et al., 2019). While studies conducted with the environmental DNA (eDNA) indicate that numerous members still remain to be identified in natural environments (e.g., deep sea and pelagic ocean; Lara et al., 2009), only two genera (i.e., Diplonema and Rhynchopus) had been described. Recent eDNA studies indicated that diplonemids are one of the most abundant and species-rich protist groups in marine environments and their ecological functions have also attracted scientific attention (Lukeš et al., 2015). After Yabuki & Tame (2015) successfully identified Hemistasia phaeocysticola, which had been incorrectly classified into Kinetoplastea, as a member of novel and independent lineage in diplonemids, another new species and genera were also reported (e.g., Tashyreva et al., 2018a; Tashyreva et al., 2018b; Prokopchuk et al. 2019). However, many diplonemids, especially those belonging to the ‘deep-sea and pelagic diplonemid (i.e., DSPD)’ clade remain to be accurately identified. Only one species belonging to the DSPD clade has been successfully described using an uncultivated sample (Okamoto et al., 2019). Further more precise identification of diplonemids including DSPD members is needed.
While DSPDs were originally shown to be distributed in deep-sea water (Lara et al., 2009), they are now considered to be widely distributed (Lukeš et al., 2015). Gawryluk et al. (2016) reported several light microscopic pictures of DPSDs; however, their cultures have not yet been established, and their ultrastructural and ecological information is still lacking. For a more precise understanding of DSPDs, the establishment of their cultures is needed. To detect the presence of diplonemid species in a sample, eDNA analysis with diplonemid specific primers would be one of the most efficient approaches. Although studies conducted with diplonemid specific primers have been previously reported (Lara et al., 2009), such approaches have yet to been conducted with next generation sequencers (NGS).
In this study, we designed primers that help to amplify diplonemid sequences specifically for NGS analysis. Applying this approach, we also analyse the diversity of diplonemids in two deep-sea water samples that were regularly pumped for commercial usage in Japan. These analyses indicate that many diplonemids, including DSPDs, are included in pumped deep-sea water and are possibly good sources from which new diplonemid species could be found.
MATERIALS AND METHODS
Water collection and filtration
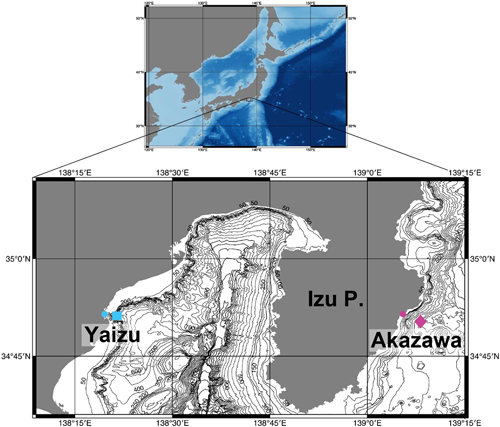
Water samples were collected at DHC deep-sea water pumping facility (Akazawa, Shizuoka) and Shizuoka prefectural deep-sea water pumping facility (Yaizu, Shizuoka) (Fig. 1). Deep-sea water was pumped off Akazawa at a depth of 800 m and off Yaizu at a depth of 397 m. The details of water samples labelled as ‘Akazawa’ and ‘Yaizu’ are summarized in Table 1. The collected water was subsequently filtered onsite through a 0.45 µm Sterivex filter (Merck, Darmstadt, Germany). After filtration, Sterivex tubes were filled with RNAlater (Life Technologies, Carlsbad, CA) and kept at −30°C until the DNA extraction.
Table 1. Summary of water and DNA samples
| Sample ID |
Collecting date |
Collecting time |
Filtered water volume |
Filtration time |
Final DNA concentration (in 200 µl) |
| Akazawa |
25, Sept. 2019 |
13:03, JST |
20 L |
39 min |
8.7 ng/µl |
| Yaizu |
26, Sept. 2019 |
13:18, JST |
10 L |
41 min |
24.9 ng/µl |
| 10 L |
41 min |
Each Sterivex tube was gently opened and the filter cut into 3 mm wide pieces. Environmental DNA was extracted from the cut Sterivex filters using DNeasy PowerSoil Kit (Qiagen, Valencia, CA) according to the manufacturer’s procedure and then purified using NucleoSpin gDNA Clean-up kit (Takara Bio, Kusatsu, Japan). The purified DNA samples were diluted in 200 µl of elution buffer. The diplonemid 18S rRNA gene sequences were amplified using Ex-Taq (Takara Bio) in two steps (i.e., 1st PCR and semi-nested PCR) with newly designed primers. The forward primer S616F_Cerco (5´-TTA AAA AGC TCG TAG TTG-3´: Fiore-Donno et al., 2018) and the reverse primer S963R_Dip (5´-ACA CTC TCG TTC TTG ATT AAT-3´: designed in this study) were utilized in the first PCR, and then S948R_Dip (5´-AAT GAA GAC ATT CTT GTC-3´: designed in this study) was utilized instead of the S963R_Dip primer in the semi-nested PCR. Both S616F_Cerco and S963R_Dip were designed in very conserved regions; therefore, the 1st PCR was expected to amplify 18S rRNA gene sequences of more diverse eukaryotes including diplonemids. Alternatively, S948R_Dip was designed to anneal to diplonemid sequences specifically. Its two bases from 3´ end were selected to not anneal to other known eukaryotic 18S rRNA gene sequences. Both PCRs were conducted with the same program: an initial denaturation step at 95°C for 2 min, 24 cycles at 95°C for 30 s, 50°C for 30 s, 72°C for 30 s; and a final extension step at 72°C for 5 min. In the 1st PCR, 1 µl of the eDNA was added to 9 µl of the PCR regent mix. The 1st PCR products were subjected to ExoSAP-IT Express PCR Cleanup Reagents (Life Technologies) and then utilized in the semi-nested PCR, which was conducted with 30 µl of PCR reagent, which included 3 µl of the template DNA solution. The amplicon library construction and a 2×300 bp pair-end sequencing by MiSeq Benchtop Sequencer (Illumina, San Diego, CA) were performed at Bioengineering Lab. Co., Ltd (Sagamihara, Japan). The raw reads were subjected to QIIME 2 and the representative sequences were reconstructed (Bolyen et al., 2019): minor abundance reads (i.e., <0.1%) were excluded and a similarity threshold of 97% was applied in sequence clustering, after the quality check (i.e., sequence trimming, quality filtering, denoising and chimera detection) with default setting and read assembly based on more than 10 bp overlap were conducted.
Phylogenetic analyses
The representative sequences were automatically aligned together with the sequences of diplonemid species by MAFFT v7.032b (Katoh et al., 2002). The alignment was checked by eye, and then sites for phylogenetic analysis were selected using trimAl v1.4. rev22 (Capella-Gutiérrez et al., 2009). A maximum-likelihood tree was obtained from this dataset using RAxML v7.2.8 (Stamatakis, 2006) with the GTR+Γ+I model and bootstrap analyses (1,000 replicates). A Bayesian analysis was run using MrBayes v3.2.1 (Ronquist et al., 2012) with the GTR+Γ+I model. One cold and three heated Markov chain Monte Carlo (MCMC) with default chain temperatures were run for 1×106 generations, sampling trees at 100-generation intervals. 2.5×105 generations were discarded as “burn-in”.
RESULTS
A total of 20 L of pumped deep-sea water was collected at Akazawa and Yaizu, respectively. From this, 8.7 ng/µl and 24.9 µg/µl of 200 µl DNA solution were acquired, respectively (Table 1). Through the PCR conducted with newly-designed specific primers and NGS analyses, 33,017 (Akazawa) and 25,137 (Yaizu) assembled reads were obtained (Table 2). The raw sequencing data was deposited in GenBank as DRX202451 (Akazawa) and DRX202452 (Yaizu). Almost all assembled reads were assigned to diplonemid sequences as BLAST top hit (99.3% of Akazawa reads and 98.8% of Yaizu reads), and 101 and 100 representative sequences were reconstructed in each analysis (Table 2). Of these, 41 sequences were shared between the two samples; 60 and 59 sequences were unique in Akazawa and Yaizu, respectively. Unique sequences accounted for 58% (Akazawa) and 48% (Yaizu) of the assembled reads, respectively (Table 2).
Table 2. Summary of sequencing analyses
| Sample ID |
# of raw reads |
# of assembled reads |
Reads assigned to diplonemids (%) |
# of representative sequences by QIIME 2.0 |
# of sample-unique sequences |
Abundance of sample-unique sequences in assembled reads |
| Akazawa |
118,952 |
33,017 |
32,799 (99.3%) |
101 |
60 |
58% |
| Yaizu |
105,368 |
25,137 |
24,834 (98.8%) |
100 |
59 |
48% |
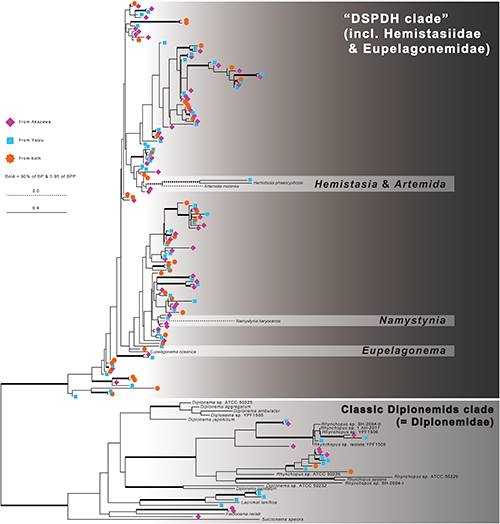
The phylogenetic analyses showed that the diplonemid sequences were divided into two sub clades: one corresponded to Diplonemidae and the others correspond to the other diplonemids, i.e. the members of Hemistasiidae, Eupelagonemidae (DSPD I) and DSPD II, collectively called DSPDH clade (Fig. 2). While the monophyly of each clade was tightly supported, the branching patterns within the clades were not resolved well. The monophyly of Hemistasiidae (i.e., Hemistasia, Artemida and Namystynia) was not recovered. The members of genus Diplonema did not form a single clade either. Of 160 representative sequences, 137 sequences fell into DSPDH clade and the other 23 sequences fell into Diplonemidae clade (Fig. 2). Both clades contain the unique sequences from Akazawa or Yaizu and the sequences derived from both. Any environmental sequences amplified in this study did not exactly match to the sequences of described species. The sequences of all described members whose 18S rRNA gene sequences were currently available were different from one another, even in the region focused in this study.
DISCUSSION
The new primer sets that were designed in this study showed highly specific amplification of diplonemid sequences (ca. 99%). Such high specificity is a big advantage in analysing the diversity of a target group using NGS. Additionally, these primer sets were able to amplify both classical diplonemids and members of the DSPDH clade. Therefore, the analysis conducted with these primer sets greatly contributes in monitoring the distribution and diversity of diplonemids. The information derived from these primer sets appears to aid in the selection of the site to collect the sample for DSPD culturing. The sequences belonging to the DSPDH clade were detected more often than those belonging to the Diplonemidae clade, which is consistent with the finding of the TARA Oceans’ study (i.e., Flegontova et al., 2016). Therefore, the DSPDs may be distributed more than the members of Diplonemidae in the deep-sea water around Izu peninsula.
While many environmental sequences were successfully and efficiently retrieved in this study, the relationships among the amplified sequences were not clearly resolved for some branches. Neither monophyly of the genus Diplonema, nor that of the family Hemistasiidae (i.e., Hemistasia, Artemida and Namystynia) were recovered in our analysis, although their monophyly was supported well in previous studies (e.g., Prokopchuk et al., 2019). This is probably due to the low phylogenetic signal caused by the short amplified region. Therefore, it is impossible to discuss the whole phylogeny of diplonemids and their branching pattern at the higher scale based on the sequences amplified here. However, since each species can be distinguished by this short-amplified region, these primers are useful to distinguish and barcode each species. Of the 160 representative sequences, 41 were commonly detected in Akazawa and Yaizu. Although further analysis is needed, the diplonemids corresponding to these 41 sequences may be distributed widely around the Izu Peninsula. The diplonemids corresponding to the other 60 sequences in Akazawa and 59 sequences in Yaizu might be distributed locally on the date when the analysed water was sampled. It is also interesting that these unique members may have occupied about half of each diplonemid community in each environment (i.e., 58% vs. 48%, Table 2). Since some well-studied species, such as Diplonema papillatum and Hemistasia phaeocysticola, behave differently in natural environments (i.e., scavenger on water plants vs. active predator on living micro algae), the ecological functions of possible unique members of Akazawa and Yaizu may be also different. If their distributions are endemic, it is quite interesting and important to understand their physiological preferences and ecological functions in discussing the environmental adaptation of microbes and characterization of two sites over Izu peninsula. For addressing these points, further studies to confirm their distribution patterns and ecological functions are needed.
This study also shows the high potential of the pumped deep-sea water to find novel diplonemids including DPSDs. Although successful attempts to find DSPDs in natural deep-sea water samples have been previously conducted (Gawryluk et al., 2016), their cultures have not yet been established. The findings of this study indicate that many DSPDs are included in easily accessible pumped deep-sea water. Therefore, it can be considered that the establishment of DSPD cultures are more highly expected using these water samples. DSPDs are very important in considering not only the diversity of microbial eukaryotes and its monitoring, but also evolution of their mitochondrial genomes. The mitochondrial genomes have been sequenced from various eukaryotes so far, but diplonemids are known to have the most eccentric structure. Diplonemid mitochondria contain numerous circular DNA molecules (minicircles) and each chromosome possesses “gene module(s)” that are a piece of the coding regions (Yabuki et al., 2016; Burger et al., 2018). Due to this eccentric structure, each module is transcribed separately from multiple minicircles and trans-splicing (e.g., uridine-insertion and RNA assembly) is followed to generate mature mRNA molecules. The evolution of such a complex mechanism is not fully understood and it is suspected that the mitochondrial genome of DSPDs may hold key information about it. If the members of DSPD are isolated from the pumped deep-sea water and studies using them are successfully conducted, the evolution of RNA-editing and RNA trans-splicing could also be better understood.
Acknowledgments
We thank Messrs. Michiyasu Nomura (DHC Corporation), Yusuke Sano and Hideo Kawai (Shizuoka prefectural deep-sea water pumping facility) for technical help with sample collection. We also thank Dr. Katsuhisa Yamada (DHC Corporation), Mr. Takahiro Amano (Shizuoka Prefectural Government Office), and Ms. Keiko Maeda (Shizuoka prefectural deep-sea water pumping facility) for logistic support to utilize pumped deep-sea water and Mr. Robert Collins for English-language corrections. This study was partially supported by a grant from the Japan Society for the Promotion of Science (17K19434) and the Institute of Fermentation, Osaka (G-2020-1-014) awarded to AY.
REFERENCES
- Adl, S. M., Bass, D., Lane, C. E., Lukeš, J., Schoch, C. L., Smirnov, A., … Cárdenas, P. (2019). Revisions to the classification, nomenclature, and diversity of eukaryotes. J. Eukaryot. Microbiol., 66, 4-119.
- Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., … Bai, Y. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol., 37, 852-857.
- Burger, G., & Valach, M. (2018). Perfection of eccentricity: mitochondrial genomes of diplonemids. IUBMB Life, 70, 1197-1206.
- Capella-Gutiérrez, S., Silla-Martínez, J. M., & Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics, 25, 1972-1973.
- Fiore-Donno, A. M., Rixen, C., Rippin, M., Glaser, K., Samolov, E., Karsten, U., … Bonkowski, M. (2018). New barcoded primers for efficient retrieval of cercozoan sequences in high-throughput environmental diversity surveys, with emphasis on worldwide biological soil crusts. Mol. Ecol. Resour., 18, 229-239.
- Flegontova, O., Flegontov, P., Malviya, S., Audic, S., Wincker, P., De Vargas, C., … Horák, A. (2016). Extreme diversity of diplonemid eukaryotes in the ocean. Curr. Biol., 26, 3060-3065.
- Gawryluk, R. M., del Campo, J., Okamoto, N., Strassert, J. F., Lukeš, J., Richards, T. A., … Keeling, P. J. (2016). Morphological identification and single-cell genomics of marine diplonemids. Curr. Biol., 26, 3053-3059.
- Katoh, K., Misawa, K., Kuma, K. I., & Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res., 30, 3059-3066.
- Lara, E., Moreira, D., Vereshchaka, A., & López-García, P. (2009). Pan-oceanic distribution of new highly diverse clades of deep-sea diplonemids. Environ. Microbiol., 11, 47-55.
- Lukeš, J., Flegontova, O., & Horák, A. (2015). Diplonemids. Curr. Biol., 25, R702-R704.
- Okamoto, N., Gawryluk, R. M., del Campo, J., Strassert, J. F., Lukeš, J., Richards, T. A., … Keeling, P. J. (2019). A revised taxonomy of diplonemids including the Eupelagonemidae n. fam. and a type species, Eupelagonema oceanica n. gen. & sp. J. Eukaryot. Microbiol., 66, 519-524.
- Prokopchuk, G., Tashyreva, D., Yabuki, A., Horák, A., Masařová, P., & Lukeš, J. (2019). Morphological, ultrastructural, motility and evolutionary characterization of two new Hemistasiidae species. Protist, 170, 259-282.
- Rognes, T., Flouri, T., Nichols, B., Quince, C., & Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ, 4, e2584.
- Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., … Huelsenbeck, J. P. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol., 61, 539-542.
- Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22, 2688-2690.
- Tashyreva, D., Prokopchuk, G., Yabuki, A., Kaur, B., Faktorová, D., Votýpka, J., … Horák, A. (2018a). Phylogeny and morphology of new diplonemids from Japan. Protist, 169, 158-179.
- Tashyreva, D., Prokopchuk, G., Votýpka, J., Yabuki, A., Horák, A., & Lukeš, J. (2018b). Life cycle, ultrastructure, and phylogeny of new diplonemids and their endosymbiotic bacteria. MBio, 9, e02447-e17.
- Yabuki, A., & Tame, A. (2015). Phylogeny and reclassification of Hemistasia phaeocysticola (Scherffel) Elbrächter & Schnepf, 1996. J. Eukaryot. Microbiol., 62, 426-429.
- Yabuki, A., Tanifuji, G., Kusaka, C., Takishita, K., & Fujikura, K. (2016). Hyper-eccentric structural genes in the mitochondrial genome of the algal parasite Hemistasia phaeocysticola. Genome Biol. Evol., 8, 2870-2878.