Materials and methods
1. Synthesis of optically active (−)-cinmethylin [(−)-1]1.1. (1S,2R)-4-Isopropyl-1-methylcyclohex-3-ene-1,2-diol (4)To a solution of AD-mix-β (40.0 g) in t-BuOH (100 mL) and H2O (100 mL) was added methanesulfonamide (2.69 g, 28.3 mmol). After being stirred at 0°C for 10 min, α-terpinene (3) (5.10 mL, 28.3 mmol) added. The mixture was stirred at 0°C for 17 hr and was added Na2SO3 (15.0 g). The resulting mixture was extracted with EtOAc three times, and the combined extracts were dried over MgSO4. After removal of solvent, the residue was purified by chromatography on silica gel (hexane/EtOAc) to give diol 4 (3.24 g, 67%): 73% ee determined by HPLC analysis of mono-MTPA ester [Chiralcel OD-H, 0.5% i-PrOH/hexane, 1.0 mL/min, tR/min=8.4 [(−)-isomer, major], 12.3 [(+)-isomer, minor]]; Rf=0.22 (hexane/EtOAc=3 : 1); [α]D22 −43.5 (c 1.01, CHCl3); IR (KBr) 3366, 2960, 1418, 1148 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.02 (d, J=6.8 Hz, 3H), 1.03 (d, J=6.8 Hz, 3H), 1.21 (s, 3H), 1.56–1.64 (m, 1H), 1.80 (ddd, J=13.2, 7.5, 5.9, 1H), 1.95–2.04 (m, 2H), 2.12–2.27 (m, 2H), 2.29 (s, 1H), 3.80 (d, J=5.2 Hz, 1H), 5.43–5.45 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 21.35, 21.42, 24.4, 24.5, 32.4, 34.5, 70.1, 71.8, 119.6, 147.8; HRMS (FD) calcd for C10H18O2 [M]+ 170.13068, found 170.13031.
1.2. (1R,2R,3S,6R)-2-[(tert-Butyldimethylsilyl)oxy]-6-isopropyl-3-methyl-7-oxabicyclo[4.1.0]heptan-3-ol (5)To a solution of diol 4 (1.37 g, 8.02 mmol) in DMF (80 mL) were added imidazole (1.37 g, 20.1 mmol) and TBSCl (2.42 g, 16.0 mmol). After being stirred at room temperature for 10 hr, the mixture was diluted with saturated NaHCO3. The resulting mixture was extracted with EtOAc three times. The combined extracts were dried over MgSO4 and concentrated to give the crude silyl ether, which was used for the next reaction without further purification.
To a solution of the above silyl ether in CH2Cl2 (80 mL) was added m-CPBA (70% in H2O, 2.37 g, 9.62 mmol). The mixture was stirred at room temperature for 5 hr and diluted with saturated NaHCO3. The resulting mixture was extracted with CH2Cl2 three times. The combined extracts were dried over MgSO4 and concentrated. The residue was purified by chromatography on silica gel (hexane/EtOAc) to give epoxide 5 (1.24 g, 51%): Rf=0.45 (hexane/EtOAc=15 : 1); [α]D19 −16.6 (c 0.994, CHCl3); IR (neat) 3565, 2961, 1259, 1094, 1078 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.13 (s, 3H), 0.18 (s, 3H), 0.955 (s, 9H), 0.957 (d, J=6.8 H, 3H), 0.98 (d, J=6.8 H, 3H), 1.09 (s, 3H), 1.22–1.34 (m, 1H), 1.47–1.56 (m, 2H), 1.64–1.72 (m, 1H), 2.04 (ddd, J=14.6, 12.4, 5.4 Hz, 1H), 2.57 (d, J=2.4 Hz, 1H), 2.63 (s, 1H), 3.54 (s, 1H); 13C NMR (100 MHz, CDCl3) δ −4.9, −4.3, 17.6, 18.1, 18.4, 18.7, 25.9 (3C), 27.9, 28.2, 34.4, 61.3, 65.3, 68.0, 73.1; HRMS (FI) calcd for C16H33O3Si1 [M+H]+ 301.21990, found 301.221933.
1.3. (1S,2R,3R,4S)-3-[(tert-Butyldimethylsilyl)oxy]-1-isopropyl-4-methyl-7-oxabicyclo[2.2.1]heptan-2-ol (6)To a solution of epoxide 5 (880 mg, 2.93 mmol) in THF (30 mL) was added p-TsOH·H2O (557 mg, 2.93 mmol). The resulting mixture was stirred at room temperature for 2.5 hr and diluted with saturated NaHCO3. The mixture was extracted with EtOAc three times, and the combined extracts were dried over MgSO4. After removal of solvent, the residue was purified by chromatography on silica gel (hexane/EtOAc) to give alcohol 6 (793 mg, 90%): Rf=0.43 (hexane/EtOAc=10 : 1); [α]D23 −9.41 (c 1.02, CHCl3): IR (neat) 3465, 1463, 1258 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.08 (s, 3H), 0.10 (s, 3H), 0.92 (s, 9H), 0.97 (d, J=6.8 Hz, 3H), 0.99 (d, J=6.8 Hz, 3H), 1.29 (s, 3H), 1.40–1.51 (m, 1H), 1.52–1.62 (m, 3H), 2.02–2.10 (m, 2H), 3.50 (s, 1H), 3.78 (s, 1H); 13C NMR (100 MHz, CDCl3) δ −4.8, −4.4, 16.9, 18.1, 18.3, 24.4, 25.9 (3C), 32.0 (2C), 34.3, 85.6, 85.7, 86.0, 89.0; HRMS (FI) calcd for C16H32O3Si1 [M]+ 300.21207 found 300.21241.
1.4. O-[(1S,3R,4S)-3-[(tert-Butyldimethylsilyl)oxy]-1-isopropyl-4-methyl-7-oxabicyclo[2.2.1]heptan-2-yl]S-methyl carbonodithioate (7)To an ice-cold solution of alcohol 6 (1.00 g, 3.33 mmol) in THF (33 mL) was added NaH (55% dispersion in mineral oil, 290 mg, 6.65 mmol). After being stirred at room temperature for 1 hr, CS2 (0.40 mL, 6.65 mmol) was added. After being stirred at room temperature for 1.5 hr, MeI (0.42 mL, 6.65 mmol) was added. The mixture was stirred at room temperature for 12 hr and concentrated. The residue was added water and then extracted with EtOAc twice. The combined extracts were concentrated. The residue was purified by chromatography on silica gel (hexane/EtOAc) to give dithioester 6 (1.26 g, 97%): Rf=0.70 (hexane/EtOAc=10 : 1); [α]D28 −4.60 (c 1.04, CHCl3); IR (neat) 2959, 1471, 1217, 1079 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.01 (s, 3H), 0.02 (s, 3H), 0.89 (s, 9H), 0.92 (d, J=6.8 Hz, 3H), 0.98 (d, J=6.8 Hz, 3H), 1.34 (s, 3H), 1.54–1.75 (m, 3H), 2.00–2.13 (m, 2H), 2.59 (s, 3H), 3.72 (d, J=2.0 Hz, 1H), 6.00 (t, J=2.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ −4.75, −4.71, 17.1, 17.66, 17.70, 18.2, 19.4, 25.8 (3C), 27.3, 31.5, 34.1, 83.0, 86.4, 89.0, 91.9, 215.0; HRMS (FI) calcd for C18H34O3S2Si1 [M]+ 390.17186, found 390.1722.
1.5. tert-Butyl[[(1S,2R,4R)-4-isopropyl-1-methyl-7-oxabicyclo[2.2.1]heptan-2-yl]oxy]dimethylsilane (8)A solution of dithioester 7 (1.24 g, 3.17 mmol), Bu3SnH (1.70 mL, 6.34 mmol), and AIBN (323 mg, 1.97 mmol) in toluene (32 mL) was refluxed for 2 hr. The resulting mixture was concentrated. The residue was purified by chromatography on silica gel (hexane/EtOAc) to silyl ether 8 (904 mg, quant): Rf=0.36 (hexane/EtOAc=25 : 1); [α]D24 −19.3 (c 1.01, CHCl3); IR (neat) 2959, 1471, 1256, 10796, 1069 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.04 (s, 3H), 0.05 (s, 3H), 0.89 (s, 9H), 0.95 (d, J=6.8 Hz, 3H), 0.96 (d, J=6.8 Hz, 3H), 1.36 (s, 3H), 1.38–1.44 (m, 3H), 1.50–1.60 (m, 2H), 1.97–2.13 (m, 2H), 3.79 (dd, J=6.8, 2.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ −4.7, −4.5, 16.7, 18.1, 18.4, 26.0 (4C), 31.4, 32.6, 33.4, 46.5, 85.8, 88.4 (2C); HRMS (FD) calcd for C16H32O2Si1 [M]+ 283.20933, found 283.21037.
1.6. (1S,2R,4R)-4-Isopropyl-1-methyl-7-oxabicyclo[2.2.1]heptan-2-ol (9)To a solution of silyl ether 8 (870 mg, 3.06 mmol) in THF (30 mL) was added TBAF (1.0 M in THF, 3.67 mL, 3.67 mmol). The solution was stirred at room temperature for 21 hr and diluted with saturated NH4Cl. The resulting mixture was extracted with EtOAc three times. The combined extracts were dried over MgSO4 and concentrated. The residue was purified by chromatography on silica gel (hexane/EtOAc) to give alcohol 8 (420 mg, 81%): Rf=0.25 (hexane/EtOAc=2 : 1); [α]D25 −0.875 (c 1.02, CHCl3); IR (neat) 3254, 2964, 1117, 1053 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.96 (s, 3H), 0.97 (s, 3H), 1.34–1.41 (m, 1H), 1.43 (s, 3H), 1.45–1.51 (m, 2H), 1.51–1.65 (m, 3H), 2.01–2.14 (m, 2H), 3.69–3.82 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 16.4, 18.1, 18.2, 32.58, 32.61, 32.9, 45.1, 76.9, 85.6, 88.7; HRMS (FI) calcd for C10H18O2 [M]+ 170.13068, found 170.13095.
1.7. (−)-Cinmethylin [(−)-1]To a solution of alcohol 8 (120 mg, 0.71 mmol) in THF (33 mL) was added NaH (55% dispersion in mineral oil, 76.8 mg, 1.76 mmol). After being stirred at room temperature for 1 hr, α-bromo-o-xylene (0.19 mL, 1.4 mmol) was added. The mixture was stirred at room temperature for 19 hr and diluted with saturated NH4Cl. The resulting mixture was extracted with (hexane/EtOAc=4 : 1) three times. The combined extracts were dried over MgSO4 and concentrated. The residue was purified by chromatography on silica gel (hexane/EtOAc) to give (−)-cinmethylin [(−)-1] (248 mg, 88%) as a colorless oil: Rf=0.76 (hexane/EtOAc=2 : 1); [α]D21 −48.8 (c 0.855, CHCl3); IR (neat) 2962, 1465, 1212, 1068, 743 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.98 (t, J=6.8 Hz, 6H), 1.40–1.47 (m, 2H), 1.48 (s, 3H), 1.50–1.68 (m, 3H), 1.95 (dd, J=6.8 Hz, 1H), 2.12 (quint, J=6.8 Hz, 1H), 2.32 (s, 3H), 3.54 (t, J=6.8 Hz, 1H), 4.36 (d, J=12.4 Hz, 1H), 4.55 (d, J=12.4 Hz, 1H), 7.12–7.22 (m, 3H), 7.30–7.35 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 16.7, 18.2, 18.3, 18.9, 32.0, 32.8, 34.0, 42.0, 68.4, 82.7, 85.5, 88.7, 125.7, 127.6, 128.5, 130.1, 136.5, 136.7; HRMS (FD) calcd for C18H26O2 [M]+ 274.19328, found 274.19393.
2. Evaluation of herbicidal activity on weeds of rice paddy fieldPlastic pots (100 cm2) were filled with paddy soil (clay loam). Water, fertilizer and soil puddling were added successively. Seeds of Echinochloa crus-galli (L.) var. formosensis (ECHCS), and Scirpus juncoides var. ohwianus (SCPJU) were sown on the soil surface. The pots were filled with water 3 cm from the rim. Each test compound was dissolved in a mixture acetone, polyoxyethylene styryl phenyl ether, and calcium dodecylbenzene sulfonate to give an emulsifiable concentrate. An amount of the water-diluted agent solutions was dropped on the water surface at 7 days after sowing for application. The dosage of the compounds was 1200 g a.i./hectare. The herbicidal effect was determined by visual observation of the treated plants in comparison with the untreated controls. The herbicidal rating score ranged from 0 (same growth as untreated) to 100 (complete kill).
Results and discussion
1. Synthesis of optically active (−)-cinmethylin [(−)-1] and its enantiomer (+)-1The synthesis of (−)-cinmethylin [(−)-1] and its enantiomer (+)-1 is shown in Scheme 1. Dihydroxylation of α-terpinene (3) with AD-mix-β afforded diol 47) in 67% yield with 73% ee, as determined by chiral HPLC analysis. Sharpless et al. reported that the desired diol was obtained at 86% ee, and our results under similar conditions showed a decrease in stereoselectivity. We tried to increase the equivalence of the ligand and to lower the reaction temperature, but the stereoselectivity did not improve. The secondary hydroxy group in 4 was then protected with TBSCl, and olefin part was subjected to epoxidation with m-CPBA to afford 5 in 51% yield. The epoxide ring opening of 5 with p-TsOH·H2O gave alcohol 6 in 90% yield. Alcohol 6 was converted to silyl ether 8 by Barton–McCombie deoxygenation.8) Deprotection of 8 with TBAF gave alcohol 9 in 81% yield. Finally, alkylation of 9 with α-bromo-o-xylene afforded (−)-cinmethylin [(−)-1] in 88% yield. Dihydroxylation of α-terpinene gave ent-4 with AD-mix-α in 77% yield and 32% ee, as determined by chiral HPLC analysis. The product ent-4 was then converted to (+)-1 using the same synthetic method described above.

Scheme 1. Synthesis of optically active (−)-cinmethylin [(−)-1] and its enantiomer (+)-1
We examined the herbicidal activity of the synthesized materials against two typical weeds found in paddy rice crops, namely, Echinochloa crus-galli (L.) var. formosensis (ECHCS) and Scirpus juncoides var. ohwianus (SCPJU), in a pot trial (Table 1). Both enantiomers of cinmethylin showed strong herbicidal activity against these weed species even at low doses. In addition, the (−)-1 showed higher herbicidal activity than the (+)-1. Cinmethylin is mainly absorbed by the roots and partly by the stems and leaves, and kills weeds by inhibiting cell division at the root growth points.9) We expect that pre-emergence (chemical treatment on seeds) will be more active than early-emergence (chemical treatment after germination), because cinmethylin is more readily absorbed by the root meristem. Next, we synthesized various cinmethylin analogs with a substituent at the C3 position using alcohol 6 as a key intermediate. The analogs were designed based on the above result that the (−)-form showed slightly higher herbicidal activity and the (−)-form showed higher enantiomeric excess. Then, Campe et al. reported the crystal structure of cinmethylin bound to FAT, and there is a space between the C3 position of cinmethylin with FAT.3) We focused on this space and devised a cinmethylin analogs with a substituent at the C3 position. Therefore, we considered that a analogs with a substituent at the C3-position could be a novel herbicide.
Table 1. Herbicidal activity of (−)-
1 and (+)-
1 | Paddy/water surface applicationa),b) |
|---|
| Pre-emergence | Early-post emergence |
|---|
| Sample | Dose of sample (g a.i./hectare) | ECHCS | SCPJU | ECHCS | SCPJU |
|---|
| cinmethylin | 300 | 100 | 100 | 100 | 100 |
| 75 | 100 | 100 | 100 | 100 |
| 19 | 100 | 90 | 100 | 70 |
| 4.8 | 100 | 80 | 85 | 40 |
| 2.4 | 100 | 40 | 40 | 0 |
| (−)-1 | 300 | 100 | 100 | 100 | 100 |
| 75 | 100 | 100 | 100 | 100 |
| 19 | 100 | 90 | 100 | 70 |
| 4.8 | 100 | 80 | 90 | 40 |
| 2.4 | 100 | 0 | 70 | 0 |
| (+)-1 | 300 | 100 | 100 | 100 | 100 |
| 75 | 100 | 90 | 100 | 90 |
| 19 | 100 | 80 | 100 | 60 |
| 4.8 | 100 | 70 | 70 | 40 |
| 2.4 | 100 | 0 | 50 | 0 |
a) Rating scale: 0 (no effect)–100 (completely effective). b) ECHCS: Echinochloa crus-galli (L.) var. formosensis; SCPJU: Scirpus juncoides var. ohwianus
The synthesis of 13 is shown in Scheme 2. Oxidation of 6 gave ketone 10 in 96% yield. The generation of the ylide from CH3PPh3Br and n-BuLi in THF, followed by the addition of ketone 10, resulted in olefin 11 (93%). Deprotection of 11 with TBAF afforded alcohol 12 in 93% yield. The analogs 13a–l were synthesized via the alkylation of 12 with corresponding benzyl bromides.

Scheme 2. Synthesis of methylene analogs 13a–l
First, we investigated the herbicidal activity of C3 methylene analogs 13a–l (Table 2), against ECHCS and SCPJU. Analogs 13a–c exhibited herbicidal activity against ECHCS when applied at a dose of 300 g a.i./hectare, but only 13a showed high herbicidal activity at a low concentration of 75 g a.i./hectare. Analogs 13a–c did not show herbicidal activity against SCPJU in the early post-emergence stage. This decreases in activity with substitution position was similar to that observed when substituents other than the methyl group (13d–i) were applied. As cinmethylin (1) also has a methyl group at the C2 position on its benzene ring, the position of the substituent on the benzene ring has a significant effect on the herbicidal activity of the product. Next, we examined the effect of the type of substituent on the herbicidal activity of analogs 13d–l. Analogs with methyl and methoxy groups, which are electron-donating groups, as well as halogens, which are electron-withdrawing groups, showed excellent herbicidal activity even at low concentrations (13a, 13d, 13g, and 13j). By contrast, analogs with slightly bulky substituents, such as trifluoromethyl (13k) and trifluoromethoxy (13l) groups, did not show herbicidal activity. Analogs 13a, 13d, 13g, and 13j and cinmethylin showed similarly high herbicidal activity against ECHCS.
Table 2. Herbicidal activity of methylene analogs
13a–
l |
|---|
| Paddy/water surface applicationa),b) |
|---|
| Pre-emergence | Early-post emergence |
|---|
| Sample | R1 | Dose of sample (g a.i./hectare) | ECHCS | SCPJU | ECHCS | SCPJU |
|---|
| (−)-1 | 2-Me | 300 | 100 | 100 | 100 | 100 |
| 75 | 100 | 100 | 100 | 100 |
| 13a | 2-Me | 300 | 100 | 100 | 100 | 0 |
| 75 | 100 | 100 | 100 | 0 |
| 13b | 3-Me | 300 | 90 | 0 | 90 | 0 |
| 75 | 0 | 0 | 0 | 0 |
| 13c | 4-Me | 300 | 100 | 90 | 100 | 0 |
| 75 | 0 | 0 | 0 | 0 |
| 13d | 2-OMe | 300 | 100 | 100 | 100 | 100 |
| 75 | 100 | 100 | 70 | 100 |
| 13e | 3-OMe | 300 | 0 | 0 | 0 | 0 |
| 75 | 0 | 0 | 0 | 0 |
| 13f | 4-OMe | 300 | 0 | 0 | 0 | 0 |
| 75 | 0 | 0 | 0 | 0 |
| 13g | 2-F | 300 | 100 | 90 | 100 | 80 |
| 75 | 100 | 90 | 100 | 70 |
| 13h | 3-F | 300 | 100 | 100 | 100 | 100 |
| 75 | 0 | 0 | 0 | 0 |
| 13i | 4-F | 300 | 100 | 0 | 100 | 0 |
| 75 | 100 | 0 | 0 | 0 |
| 13j | 2-Cl | 300 | 100 | 100 | 100 | 70 |
| 75 | 100 | 90 | 100 | 70 |
| 13k | 2-CF3 | 300 | 0 | 0 | 0 | 0 |
| 75 | 0 | 0 | 0 | 0 |
| 13l | 2-OCF3 | 300 | 0 | 0 | 0 | 0 |
| 75 | 0 | 0 | 0 | 0 |
a) Rating scale: 0 (no effect)–100 (completely effective). b) ECHCS: Echinochloa crus-galli (L.) var. formosensis; SCPJU: Scirpus juncoides var. ohwianus
Next, we synthesized a novel cinmethylin analogs with a substituent at the C3 position (Scheme 3). Alkylation of alcohol 6, followed by deprotection with TBAF, afforded alcohol 15. Alkylation of 15 with α-bromo-o-xylene resulted in the synthesis of 16a in 71% yield. Then, protection of alcohol 6 with ethyl vinyl ether/PPTS, followed by deprotection with TBAF, afforded alcohol 18 in good yield. Alkylation of alcohol 18 with α-bromo-o-xylene afforded ether 19a in 94% yield. Ether 19a was converted to alcohol 20a under acidic conditions in 89% yield. Alcohol 20a was oxidized with PCC to give 21a in 67% yield. Finally, ketone 21a was converted to oxime 22a using NH2OH·HCl/NaOAc in 67% yield.

Scheme 3. Synthesis of analogs 16a, 20a–22a
The synthesis of methyl analog 24a with a methyl group at the C3 position is shown in Scheme 4. Reduction of olefin 12 and subsequent alkylation of the alcohol gave 24a. 1H NMR analysis indicated that the stereoisomeric ratio of 24a at the C3 position was 44 : 56. Thus, analog 24a was as such subjected to the herbicidal activity test.

Scheme 4. Synthesis of methyl analog 24a
The herbicidal activity of the cinmethylin analogs with various substituents at the C3 position are shown in Table 3. The carbonyl analog 21a and oxime analog 22a showed excellent activity against ECHCS, even at low concentrations, similar to (−)-1 and the methylene analog 13a. The herbicidal activity of methoxy analog 16a and hydroxy analog 20a was evident at 300 g a.i./hectare but decreased or disappeared at 75 g a.i./hectare. The methyl analog 24a showed high activity even at low concentrations. These results suggest that the substituent at the C3 position affects the herbicidal activity of the resultant compound. Studies on the relationship between the type of substituent at the C3 position and herbicidal activity of the cinmethylin analog are ongoing in our laboratory.
Table 3. Herbicidal activity of analogs
13a–
24a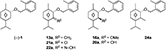 |
|---|
| Paddy/water surface applicationa),b) |
|---|
| Pre-emergence | Early-post emergence |
|---|
| Sample | Dose of sample (g a.i./hectare) | ECHCS | SCPJU | ECHCS | SCPJU |
|---|
| (−)-1 | 300 | 100 | 100 | 100 | 100 |
| 75 | 100 | 100 | 100 | 100 |
| 13a | 300 | 100 | 100 | 100 | 0 |
| 75 | 100 | 100 | 100 | 0 |
| 21a | 300 | 100 | 100 | 100 | 0 |
| 75 | 100 | 80 | 100 | 0 |
| 22a | 300 | 100 | 100 | 100 | 100 |
| 75 | 100 | 100 | 100 | 70 |
| 16a | 300 | 90 | 90 | 90 | 40 |
| 75 | 0 | 0 | 0 | 0 |
| 20a | 300 | 90 | 100 | 100 | 80 |
| 75 | 40 | 40 | 0 | 0 |
| 24a | 300 | 90 | 90 | 90 | 90 |
| 75 | 90 | 90 | 90 | 50 |
a) Rating scale: 0 (no effect)–100 (completely effective). b) ECHCS: Echinochloa crus-galli (L.) var. formosensis; SCPJU: Scirpus juncoides var. ohwianus


