Abstract
Superior plant architecture is a key means of enhancing yield potential in high yielding varieties. A newly identified recessive gene, named sd-c, controls plant height and tiller number. Genetic analysis of an F2 population from a cross between the semi-dwarf mutant and japonica cv. Houshengheng showed that the sd-c locus was flanked by SSR markers RM27877 and RM277 on chromosome 12. Thirty nine InDel markers were developed in the region and the sd-c gene was further mapped to a 1 cM centromeric region between InDel markers C11 and C12. These sequenced markers can be used to distinguish wild type and mutants and thus can be used in marker-assisted selection. The sd-c mutant decreases culm length by about 26% and doubles the tiller number without changing seed weight. Until now only sd-1 has been used in indica rice breeding programs. The sd-c mutant seems to have no undesirable pleiotropic effects and is therefore a potential genetic resource for breeding semi-dwarf indica rice cultivars.
Introduction
Ideal plant type in crops has become a significant objective in recent years (Ji et al. 2006). Semi-dwarfism and uniform tillering capacity are important components of those ideo-types in most cereal crops, including rice. The genes underlying dwarfism and semi-dwarfism affect various physiological and biochemical properties and therefore cloning and functional analyses could improve our ability to utilize them in high yield rice production (Gao et al. 2005, Ma et al. 2009).
The widely used semi-dwarf genes sd-1 in rice and Rht1 and Rht2 in wheat are known as the “Green Revolution” genes (Hedden 2003). These genes are involved in endogenous gibberellin response and their presence lead to shorter and thicker culms, resulting in higher harvest index and lodging resistance that permits greater responsiveness to nitrogen fertilizer (Lu et al. 1979, Xiong et al. 1988). These genes contributed to significant worldwide increases in crop production from the 1960s and 1970s.
The Sd-1 allele is the only semi-dwarfing gene used in indica rice. However, it is important not to rely on a single gene for any important trait, including plant height. For this reason we need to seek more sources of dwarfism and semi-dwarfism for practical breeding.
The availability of the whole genome sequences of the indica cultivar (cv.) 93-11 and japonica cv. Nipponbare makes it easy to isolate useful genes in rice by map-based cloning. More than 60 dwarf and semi-dwarf genes have been reported and cloned in rice. Several lines of evidence show that many of the genes involved in reduced height and tiller number in rice are involved in the GA (gibberellin), BR (brassinosteroid) and/or strigolactone pathways (Arite et al. 2007, Itoh et al. 2004, Lin et al. 2009, Sasaki et al. 2003, Ueguchi-Tanaka et al. 2000).
Strigolactones are recently discovered hormones that control shoot branching, and particularly suppression of outgrowths from preformed axillary buds. New branching mutants have been identified with reduced strigolactone levels or with defective strigolactone regulation or response. Genes D3, D10, D14, D17 and D27 are all associated with biosynthesis of, or sensitivity to, strigolactone (Arite et al. 2007, 2009, Lin et al. 2009, Zou et al. 2005). Auxin signaling mutants have been useful in demonstrating that strigolactone levels are mediated by the classical auxin signal transduction pathway (Beveridge et al. 2010).
Early physiological work suggested that auxin depletion was responsible for branching after decapitation, but a second messenger was required for bud formation (Dun et al. 2009, Leyser 2009). Auxin depletion causes a substantial drop in the expression of strigolactone biosynthesis genes in rice and Arabidopsis (Bennett et al. 2006, Hayward et al. 2009).
Here we report a monogenic recessive mutant (sd-c) in indica rice cv. 93-11. This mutant exhibits semi-dwarf stature and high tiller number. We performed morphological observations and genetic analysis on the mutant and initiated fine mapping of the sd-c gene, with the ultimate objective of cloning and functional analysis.
Materials and Methods
Plant materials and genetic analysis
The sd-c mutant was identied as a recessive semi-dwarf spontaneous mutant in indica cv. 93-11. An initial F2 mapping population derived from a cross between sd-c and japonica cv. Houshengheng (HSS) was used to determine gene action in the mutant. The parents and 1, 660 F2 individuals were planted in 2009 in Nanjing, Jiangsu province. Another F2 population containing 50, 000 individuals was grown in Hainan province in the winter of 2010.
Hormone treatment
For analysis of hormone response the sd-c mutant and 93-11 were grown in hydroponic culture under controlled conditions (13 h light, 11 h dark, 28°C) after germination. After two weeks, GR24 which is a kind of synthetic strigolactone analog (10 μM) was added in the hydroponic medium, sd-c and 93-11 were continuously treated for one month and with water as check. To check the function of GR24, we used the d27 as a positive control. For RNA isolation and expression analysis two weeks seedlings of 93-11 and sd-c mutant were harvested and immediately frozen in liquid nitrogen. All the data were from three independent experiments technically.
Genetic analysis
The sd-c mutant was crossed with 93-11 and HSS. The segregation of normal and mutant plants in the F2 population was investigated 15 days after the heading stage, and the segregation ratio was tested by chi-squared analysis.
Molecular markers and gene mapping
SSR markers were selected for a survey of polymorphism between the, sd-c mutant and HSS; polymorphic markers were then used for linkage analysis based on semi-dwarf segregants from the F2 population. New InDel markers were identified in the region covering the target gene by aligning the genome sequences of Nipponbare and 93-11 (http://www.gramene.org/). PCR amplifications were performed as described by Panaud et al. (Panaud et al. 1996). PCR products were separated in 3% agarose gels and visualized by staining with ethidum bromide. Rice genomic DNA was isolated following the method of McCouch et al. (McCouch et al. 1988). The F2 population of sd-c × HSS was used for gene mapping. SSR markers showing polymorphism between sd-c and HSS were selected for linkage analysis using Mapmaker/Exp 3.0 (Lander et al.1987). The parents and 10 random semi-dwarf F2 plants were used for bulked segregant analysis. The linked markers were used to genotype individual semi-dwarf plants in the entire F2 population and in gene mapping.
qRT-PCR analysis
Total RNA was extracted from individual rice plants as described previously (Wadsworth et al. 1988). Complementary DNA was synthesized from total RNA using a reverse transcriptional kit from Tiangen (http://www.tiangen.com/). qRT-PCR primers used for quantitative Real-Time PCR (Table 1) were designed to flank introns. qRT-PCR was replicated at least three times and the reactions were performed on a quantitative PCR 7900HT instrument from Applied Biosystems (http://www.appliedbiosystems.com/).
Table 1
Primers used in quantitative RT-PCR and markers used for fine mapping of the
sd-c
| Genes |
|
Accession no. |
Forward primers (5′-3′) |
Rreverse primers (5′-3′) |
| D3 |
|
AK065478 |
CTCTAGAAGGAGTAGATTAG |
GAAGAGAAACCAGGGAAAAC |
| D14 |
|
AK070827 |
GTGCTGTCGCATGGCTTC |
GCAGGTCGTCGACGTAGG |
| D10 |
|
NM_001050764 |
GGTAGCAACGAGAGGCAGTT |
TCGACCTTGGTGAGCGTGTT |
| D17 |
|
AK109771 |
GATGGTGGCTATGTTCTTCT |
TAGTTATTTGGTTCCCCTGAT |
| D27 |
|
FJ641055 |
GGCTAAAGAATGAAAAGGA |
AGAGCTTGGGTCACAATCTCG |
| IAA1 |
|
AK100800 |
ACCAAGAGCCGCTCAATGAG |
ATCACACGTGGGCGAACATC |
| IAA20 |
|
AK102541 |
ACGTGAACGGGATTATTTTG |
GCTTATGAAATTGCTGAAACA |
| YUCCA6 |
|
BAC80117 |
TTCCCAGATGGTTGGAAGG |
TGTTGCGCCTCAAGATATTTG |
| OsPIN2 |
|
AK101191 |
TCAGATGCAGGGCTAGGAA |
CCACAAGAAATGATCTTTGG |
|
Actin |
|
TCATGAAGATCCTGACGGAG |
AACAGCTCCTCTTGGCTTAG |
|
Markers |
|
|
|
|
C2 |
|
AAGTTAGAGCGTAGCCAGTG |
AGTACATTTCCATCGAGTGTG |
|
C3 |
|
ATTGCAGATTACCCCTTTG |
TGTTGATGCGGACATTAGT |
|
C4 |
|
CGGGTCGATAACAAAAGCA |
GCCTAACCCACCATCTTGC |
|
C8 |
|
AAACTAAGCCAAGCCAACTC |
GGAGAAGGACAAGAGGAATG |
|
C11 |
|
TTACTCACTTCTTGCCTCTA |
CTCCTTTGTTTCGCTATT |
|
C12 |
|
TTTAATTTAATAGCACCACG |
TCCCCTTATGTCCTCTGT |
|
C21 |
|
GAGATTGATACAGGCTACA |
CGGTTGTCAGTTTTGGT |
|
C25 |
|
AAAGCCGACATAACAGC |
GGAAATGGTATCGTGCA |
|
C26 |
|
CTCCGCTCCTCCTCCTCGTC |
CCGCCTATTGTCCATCTCCA |
|
C30 |
|
CGATGATGCTTCCCCTTGC |
TGAACCCGAGCTGCCTGTG |
|
C33 |
|
TCAATGTGGAGGGAAAT |
TGATGCGGTGGTAGAAA |
Results
Phenotype of the sd-c mutant
The mutant used in this study was originally isolated as a dwarf variant in cv. 93-11 during breeding activities in our laboratory. It behaved as a recessive multi-tillered semi-dwarf mutant showing normal vegetative growth. Its phenotype at the seedling stage was similar to that of the wild type. From the tilling stage, the mutant was obviously dwarfed and multi-tillered. At maturation stage, 93-11 was 134.41 ± 5.74 cm high whereas sd-c was 99.39 ± 2.92. Each internode of the mutant had a relatively consistent reduction in length (Fig. 1); the mean tiller number of the mutant was 17 compared to 8 for 93-11. The panicle length of the mutant was only slightly shorter than that of the wild type and the 1,000 seed weight was similar (Fig. 2 and Table 2).

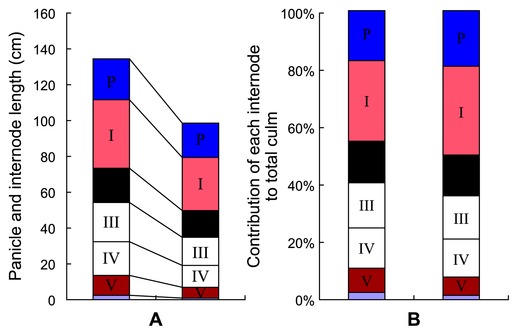
Table 2
Comparison of plant traits in 93-11 and
sd-c mutant
| Trait |
93-11 |
sd-c mutant |
P value (t test) |
| Plant height (cm) |
134.41 ± 5.74 |
99.39 ± 2.92 |
2.17 × 10−3 |
| Panicle length (cm) |
22.84 ± 0.88 |
19.12 ± 0.57 |
1.44 × 10−9 |
| First internode length (cm) |
32.97 ± 1.40 |
25.73 ± 1.22 |
3.3 × 10−10 |
| Second internode length (cm) |
15.63 ± 0.83 |
10.47 ± 0.93 |
1.27 × 10−10 |
| Third internode length (cm) |
18.28 ± 0.80 |
12.18 ± 0.46 |
4.23 × 10−14 |
| Forth internode length (cm) |
16.9 ± 0.78 |
10.77 ± 0.97 |
6.94 × 10−12 |
| Fifth internode length (cm) |
9.84 ± 0.85 |
4.93 ± 0.18 |
1.49 × 10−8 |
| Tiller number |
8 ± 1.6 |
17 ± 2.5 |
5.82 × 10−17 |
| 1000 seeds weight(g) |
20.78 ± 0.13 |
20.64 ± 0.21 |
0.601 |
| Gain yield(kg/ha) |
8573.36 ± 8.53 |
9642.79 ± 5.82 |
8.63 × 10−19 |
| Spikelet fertility |
93.21 ± 1.36 |
92.82 ± 2.36 |
0.682 |
| Number of filled grain |
152 ± 3.64 |
147 ± 4.38 |
6.16 × 10−3 |
Values are the mean ± SD of ten biological replicates.
To analyze whether the semi-dwarf phenotype was controlled by a single gene, the sd-c mutant was crossed to both 93-11 and HSS. F1 plants of both crosses were normal in height indicating recessiveness of semi-dwarf stature. In the two F2 populations, the mutant type was easy to distinguish because of the semi-dwarf and excessive tillers phenotype. In sd-c mutant × 93-11, 837 F2 plants were similar in height to the wild type and 277 were semi-dwarf, indicating segregation at a single locus (x23:1 = 0.011, P > 0.05). Similarly, the F2 population from sd-c mutant × HSS segregated 1, 247 plants phenotypically wild-type and 413 were semi-dwarf accompany with excessive tillers, again a 3 : 1 ratio (x2 = 0.013, P > 0.05) (Supplemental Fig. 1).
To elucidate the mechanism of reduced height and increased tiller numbers in sd-c, we sought to identify the sd-c gene by positional cloning. A total of 621 RM-series SSR markers were used to screen the sd-c mutant, HSS and 10 semi-dwarf F2 plants. Four SSR markers on chromosome 12, RM27877, RM7102, RM277, RM1246, co-segregated with sd-c thus suggesting linkage. Four hundred and thirteen semi-dwarf F2 plants were individually analyzed with the 4 markers. The sd-c gene was mapped to a 12.9 cM interval flanked by RM27877 and RM277 (Fig. 3A).
Fine mapping of sd-c
To fine-map the sd-c locus, a large F2 population of sd-c mutant × HSS was generated. For positional cloning it was necessary to obtain multiple plants that were recombinant in the candidate region of the target gene. The molecular markers RM27877 and RM277 were used as selection markers for large-scale mapping. We extracted DNA from 9,780 semi-dwarf plants at the tillering stage and surveyed them for RM27877 and RM277 alleles; 679 recombinant plants were identified.
The sequences between RM27877 and RM277 in the parents were then searched for new markers and 39 new InDel markers were developed in the region; 23 were polymorphic between the two parents.
These markers were used to analyze the recombinant plants in the F2 population. We found 11 and 37 recombinant plants between RM27877 and C33 and between C26 and RM277, respectively (Fig. 3B). The sd-c gene was further anchored between markers C11 (on BAC/PAC clone OSJNBa0012G19) and C12 (on BAC/PAC clone OSJNBa0017G24), which are located in the chromosome 12 centromere region. According to the Nipponbare genomic sequence the physical length of the 1 cM distance between C11 and C12 is 1,635 kb (Fig. 3C).
Hormone responses and RT-PCR analysis
The semi-dwarf stature and high tillering capacity of sd-c was similar to those of strigolactone-deficient and strigolactone-insensitive mutants; consequently, we suspected that sd-c is deficient in strigolactone biosynthesis or sensitivity. To determine whether sd-c is strigolactone-deficient or strigolactone-insensitive, we treated sd-c with the GR24. When sd-c was treated with 10 μM GR24 for one month, the semi-dwarf stature and high tillering capacity of sd-c was the same as the sd-c check but d27 showed an obvious tilling decrease. This result indicates the sd-c is likely to be a strigolactone-insensitive mutant.
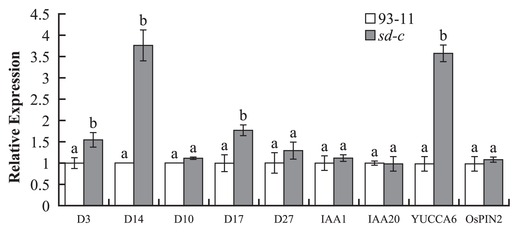
The sd-c mutant is insensitive for GR24, so the expression analysis of genes involved in the strigolactone synthesis and responses pathway in sd-c mutant were investigated by real-time PCR experiment. And also there was evidence suggesting that strigolactone contributes to plant architecture by having an important influence on auxin polar transport. Therefore, we investigated the expression levels of 5 genes affecting strigolactone and 4 genes affecting auxin metabolism (Fig. 4).
The results showed that the expression of the strigolactone synthesis genes D10, D27 and auxin synthesis genes IAA1, IAA20 were not changed, while the strigolactone responses gene D3 and D14 as well as auxin synthesis gene YUCCA6 significantly changed (Fig. 4). Surprisingly, the strigolactone synthesis genes D17 showed a significantly changed while the auxin responses gene OsPIN2 showed no change (Fig. 4). Our data demonstrated that sd-c may be involved in the strigolactone pathway.
Discussion
Rice is the most important crop in the world, providing food for more than half of the population. Increasing crop yield is a major challenge for modern agriculture. Crops with appropriate plant architectures are able to produce higher grain yields. Therefore, understanding the mechanisms that underly plant architecture will not only permit fundamental issues in plant science to be addressed, but may also facilitate the breeding of high yielding crops (Wang et al. 2008).
Rice plant architecture is crucial for grain yield and is determined by plant height, tiller number and angle and panicle morphology. Semi-dwarfism is an important trait for improving lodging resistance (Miura et al. 2009). Increased tiller numbers with no decrease in seed weight is also a useful trait for breeders. More than 60 genes affecting plant architecture have been identified (mainly in japonica rice), but most of these genes have no practical value (Liang et al. 2004). Gene sd-1 is the only semi-dwarf gene used in rice breeding. To avoid dependence on a single semi-dwarfing gene in rice we conducted further assessments of the sd-c gene by attempting to elucidate the molecular mechanisms regulating plant height and tiller number in the mutant. Since the sd-c seems to have no undesirable pleiotropic effects and is therefore a potential genetic resource for breeding semi-dwarf indica rice cultivars.
Studies on cloned genes for dwarfism and semi-dwarfism in rice have shown that reduced height is caused by deficiencies or modifications in endogenous plant hormones, such as gibberellins, brassinosteroids and strigolactone, or in their signaling pathways. Genes sd-1, d1, d18 and d35 encoding key enzymes in biosynthesis of GAs or modifications of their signaling pathway have been cloned. Genes D2, D11 and D61 participate in Br biosynthesis and signal transduction, whereas D3, D14, D10, D17 and D27 take part in strigolactone biosynthesis and signal transduction. Now, we are sequencing the region in order to find the gene and illustrate the mechanism. Further work on the sd-c mutant may provide more details on the relationship between semi-dwarfism and plant hormones (Ishikawa et al. 2005).
The sequenced rice genome has greatly facilitated the application of map-based cloning. However, near-centromere regions are still difficult to clone due to the abundance long satellite repeats and retrotransposons (Zhu et al. 2007). The sd-c locus was fine mapped to a 1, 635 kb centromeric region on chromosome 12 using 23 new InDel markers, providing essential information for the final isolation of this gene.
Mutants for various traits in rice form the basis of genetic analysis and functional genomics. Identification of genes controlling plant architecture has become important for modern rice genetics and breeders. The China National Rice Research Institute has generated and identified at least 60 mutants affecting plant architecture in rice. One of these is sd-c and the current fine mapping of the gene involved paves the way for cloning and functional analysis.
Acknowledgments
This research is supported by the grants from the Unite of National Natural Science Foundation and Yunan Province (U1036605), National Transform Science and Technology Program (2009ZX08009-107B), 863 program (2012AA101101, 2011AA10A101), Jiangsu Science and Technology Development Program (BK2010016, BE2011302).
Literature Cited
- Arite, T., H. Iwata, K. Ohshima, M. Maekawa, M. Nakajima, M. Kojima, H. Sakakibara and J. Kyozuka (2007) DWARF10, an RMS1/MAX4/DAD1 ortholog, controls lateral bud outgrowth in rice. Plant J. 51: 1019–1029.
- Arite, T., M. Umehara, S. Ishikawa, A. Hanada, M. Maekawa, S. Yamaguchi and J. Kyozuka (2009) D14, a strigolactone-insensitive mutant of rice, shows an accelerated outgrowth of tillers. Plant Cell Physiol. 50: 1416–1424.
- Bennett, T., T. Sieberer, B. Willett, J. Booker, C. Luschnig and O. Leyser (2006) The Arabidopsis MAX pathway controls shoot branching by regulating auxin transport. Current Biol. 16: 553–563.
- Beveridge, C.A. and J. Kyozuka (2010) New genes in the strigolactone-related shoot branching pathway. Curr. Opin. Plant Biol. 13: 34–39.
- Dun, E.A., P.B. Brewer and C.A. Beveridge (2009) Strigolactones: discovery of the elusive shoot branching hormone. Trends in Plant Sci. 14: 364–372.
- Gao, F.M., Y. Jiang, D.W. Kong and S.G. Li (2005) Genetic control of plant height and its utilization in rice. Mol. Plant Breed. 3: 87–93.
- Hayward, A., P. Stirnberg, C. Beveridge and L. Ottoline (2009) Interactions between auxin and strigolactone in shoot branching control. Plant Physiol. 151: 400–412.
- Hedden, P. (2003) The genes of the green revolution. Trends Genet. 19: 5–9.
- Ishikawa, S., M. Maekawa, T. Arite, K. Onishi, I. Takamure and J. Kyozuka (2005) Suppression of tiller bud activity in tillering dwarf mutants of rice. Plant Cell Physiol. 46: 79–86.
- Itoh, H., T. Tatsumi, T. Sakamoto, K. Otomo, T. Toyomasu, H. Kitano, M. Ashikari, S. Ichihara and M. Matsuoka (2004) A rice semi-dwarf gene, Tan-Ginbozu (D35), encodes gibberellin biosynthesis enzyme, ent-kaurene oxidase. Plant Mol Biol. 54: 533–547.
- Ji, Y., M.M. Miao and X.H. Chen (2006) Progresses on the molecular genetics of dwarf character in plants. Mol. Plant Breed. 4: 753–771.
- Lander, E.S., P. Green, J. Abrahamson, A. Barlow, M.J. Daly, S.E. Lincoln and L.A. Newberg (1987) MAPMAKER: An interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1: 174–181.
- Leyser, O. (2009) The control of shoot branching: an example of plant information processing. Plant Cell Environ. 32: 694–703.
- Liang, G.H., X.Y. Cao, J.M. Sui, X.Q. Zhao, C.J. Yan, C.D. Yi and M.H. Gu (2004) Fine mapping of a semidwarf gene sd-g in indica rice (Oryza sativa L.). Chinese Sci. Bull. 49: 900–904.
- Lin, H., R.X. Wang, Q. Qian, M.X. Yan, X.B. Meng, Z.M. Fu, C.Y. Yan, B. Jiang, Z. Su, J.Y. Li et al. (2009) DWARF27, an iron-containing protein required for the biosynthesis of strigolactones, regulates rice tiller bud outgrowth. Plant Cell 21: 1512–1525.
- Lu, Y.G., S.X. Zeng, Z.B. Li and K.H. Wang (1979) A study on the phenotypic expression of dwarf gene sources of early Hsien rice in China. Acta Genetica Sin. 6: 311–320.
- Ma, L.Y., J.S. Bao, X.M. Li, X.D. Zhu, Z.J. Ji, Y.W. Xia and C.D. Yang (2009) Progress on cloning and functional analysis of dwarfism related genes in rice. Chinese J. Rice Sci. 23: 1–11.
- McCouch, S.R., G. Kochert and Z.H. Yu (1988) Molecular mapping of rice chromosomes. Theor. Appl. Genet. 76: 148–159.
- Miura, K., J. Wu, H. Sunohara, X. Wu, T. Matsumoto, M. Matsumoto, M. Ashikari and H. Kitano (2009) High-resolution mapping revealed a 1.3-Mbp genomic inversion in Ssi1, a dominant semi-dwarf gene in rice (Oryza sativa). Plant Breed. 128: 63–69.
- Panaud, O.X., S.R. Chen and S.R. McCouch (1996) Development of micro satellite makers and characterization of simple sequence length polymorphisms (SSLPs) in rice (Oryza sativa L.). Mol. Gen. Genet. 252: 597–607.
- Sasaki, A., H. Itoh, K. Gomi, M. Ueguchi-Tanaka, K. Ishiyama, M. Kobayashi, D.H. Jeong, G. An, H. Kitano, M. Ashikari et al. (2003) Accumulation of phosphorylated repressor for gibberellin signaling in an F-box mutant. Science 299: 1896–1898.
- Ueguchi-Tanaka, M., Y. Fujisawa, M. Kobayashi, M. Ashikari, Y. Iwasaki, H. Kitano and M. Matsuoka (2000) Rice dwarf mutant d1, which is defective in the α subunit of the heterotrimeric G protein, affects gibberellin signal transduction. Proc Natl Acad Sci USA 97: 11638–11643.
- Wadsworth, G.J., M.G. Redinbaugh and J.G. Scandalios (1988) A procedure for the small-scale isolation of plant RNA suitable for RNA blot analysis. Anal. Biochem. 172: 279–283.
- Wang, Y.H. and J.Y. Li (2008) Molecular basis of plant architecture. Annu. Rev. Plant Biol. 59: 253–279.
- Xiong, Z.M., S.K. Min and S.H. Cheng (1988) Genetic analysis and utilization of the dwarf resources in China. Rice Rev. Abstr. 7: 1–5.
- Zhu, L., W.Z. Liu, C. Wu, W.J. Luan, Y.P. Fu, G.C. Hu, H.M. Si and Z.X. Sun (2007) Identification and fine mapping of a gene related to pale green leaf phenotype near the centromere region in rice (Oryza sativa). Rice Sci. 14: 172–180.
- Zou, J.H., Z.X. Chen, S.Y. Zhang, W.P. Zhang, G.H. Jiang, X.F. Zhao, W.X. Zhai, X.B. Pan and L.H. Zhu (2005) Characterizations and fine mapping of a mutant gene for high tillering and dwarf in rice (Oryza sativa L.). Planta 222: 604–612.