Abstract
Barley (Hordeum vulgare) is one of the world’s most important cereal crops. Although its large and complex genome has held back barley genomics for quite a while, the whole genome sequence was released in 2012 by the International Barley Genome Sequencing Consortium (IBSC). Moreover, more than 30,000 barley full-length cDNAs (FLcDNAs) are now available in the public domain. Here we present the Barley Gene Expression Database (bex-db: http://barleyflc.dna.affrc.go.jp/bexdb/index.html) as a repository of transcriptome data including the sequences and the expression profiles of barley genes resulting from microarray analysis. In addition to FLcDNA sequences, bex-db also contains partial sequences of more than 309,000 novel expressed sequence tags (ESTs). Users can browse the data via keyword, sequence homology and expression profile search options. A genome browser was also developed to display the chromosomal locations of barley FLcDNAs and wheat (Triticum aestivum) transcripts as well as Aegilops tauschii gene models on the IBSC genome sequence for future comparative analysis of orthologs among Triticeae species. The bex-db should provide a useful resource for further genomics studies and development of genome-based tools to enhance the progress of the genetic improvement of cereal crops.
Introduction
Among the major cereal crops, barley (Hordeum vulgare) is ranked fourth in worldwide production behind wheat, rice and maize (http://faostat.fao.org). However, the large genome size of about 5.1 gigabases (Gb) and the high rate of repetitive elements (>80%) of this important crop have largely hindered the development of genomic studies for many years. Ahead of barley genome sequencing, large-scale analysis of full-length cDNAs (FLcDNAs) derived from the Japanese malting barley variety ‘Haruna Nijo’ was conducted in Japan (Matsumoto et al. 2011, Sato et al. 2009). An accompanying database was also developed to provide access to the clones and sequence information. The FLcDNA information accelerates molecular and evolutionary studies, particularly of barley genes such as the CONSTANS-like (COL) gene family, which is known to control flowering (Cockram et al. 2012, Kikuchi et al. 2012). In 2012, IBSC released the whole genome sequence of barley obtained from a malting barley variety ‘Morex’ (The International Barley Genome Sequencing Consortium 2012). This provides an extensive opportunity for more comprehensive characterization of the genomic sequences on each chromosome, and insights into the overall structure and function of the entire genome. Barley has become a model organism for understanding the structure and function of Triticeae genomes and developing genomics tools for future improvement of crops.
We have upgraded the barley FLcDNA database to provide a more robust repository of information on barley gene structures and gene functions. The bex-db currently contains all data information including the sequence and annotation of FLcDNAs, the gene expression profiles based on microarray analysis using various experimental materials and conditions and the sequences of novel ESTs of barley. Here, we report the important contents and major features of our upgraded database with the detailed demonstration of various options for data access.
Database contents
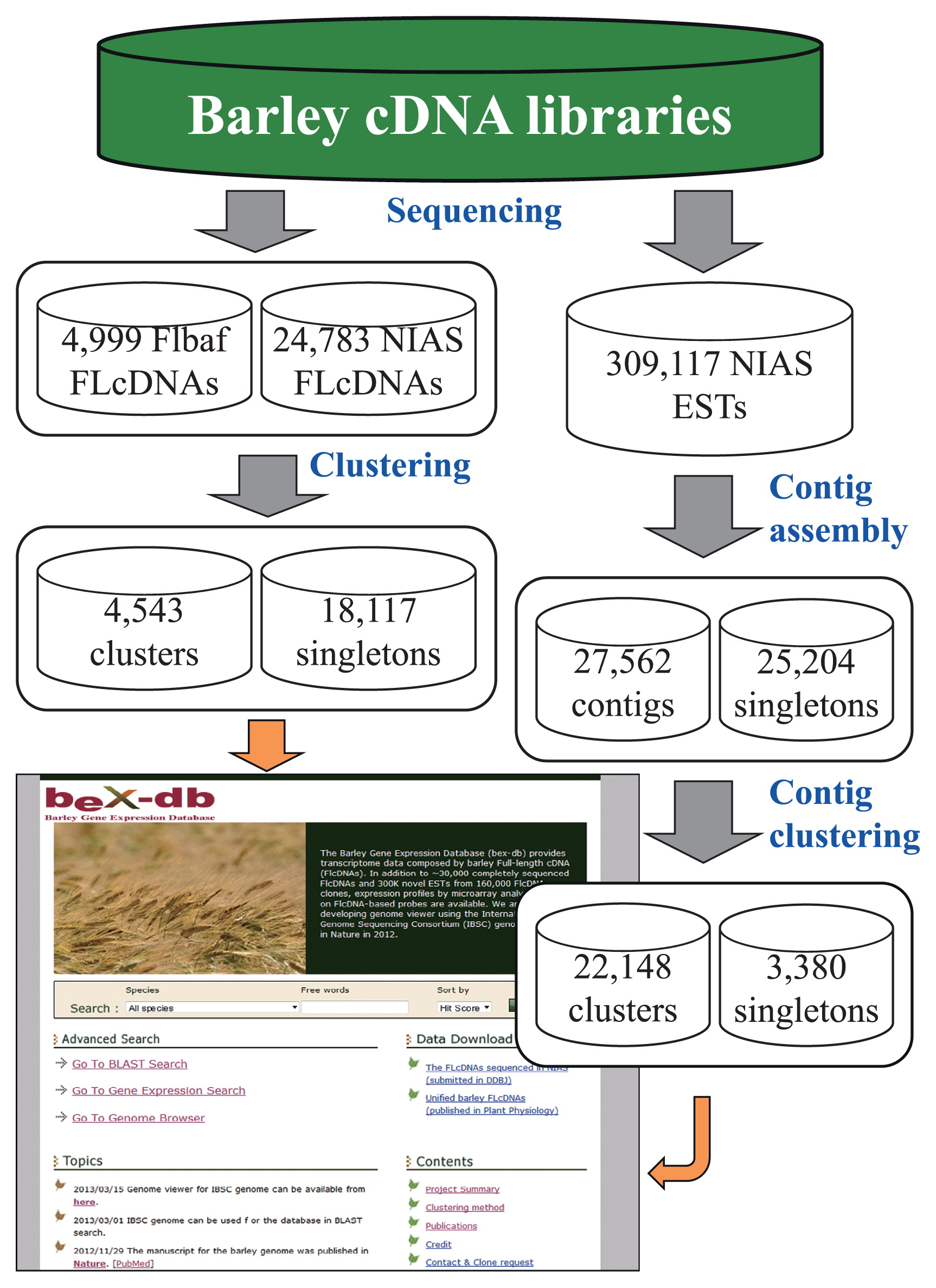
The bex-db currently contains data on nucleotide sequences, microarray gene expression profiles and structural features of expressed genes in barley (Fig. 1). The nucleotide sequence data, which all contain clone IDs, include full-length sequences (FLcDNA) or partial length sequences of clones corresponding to the ESTs.
Nucleotide sequences
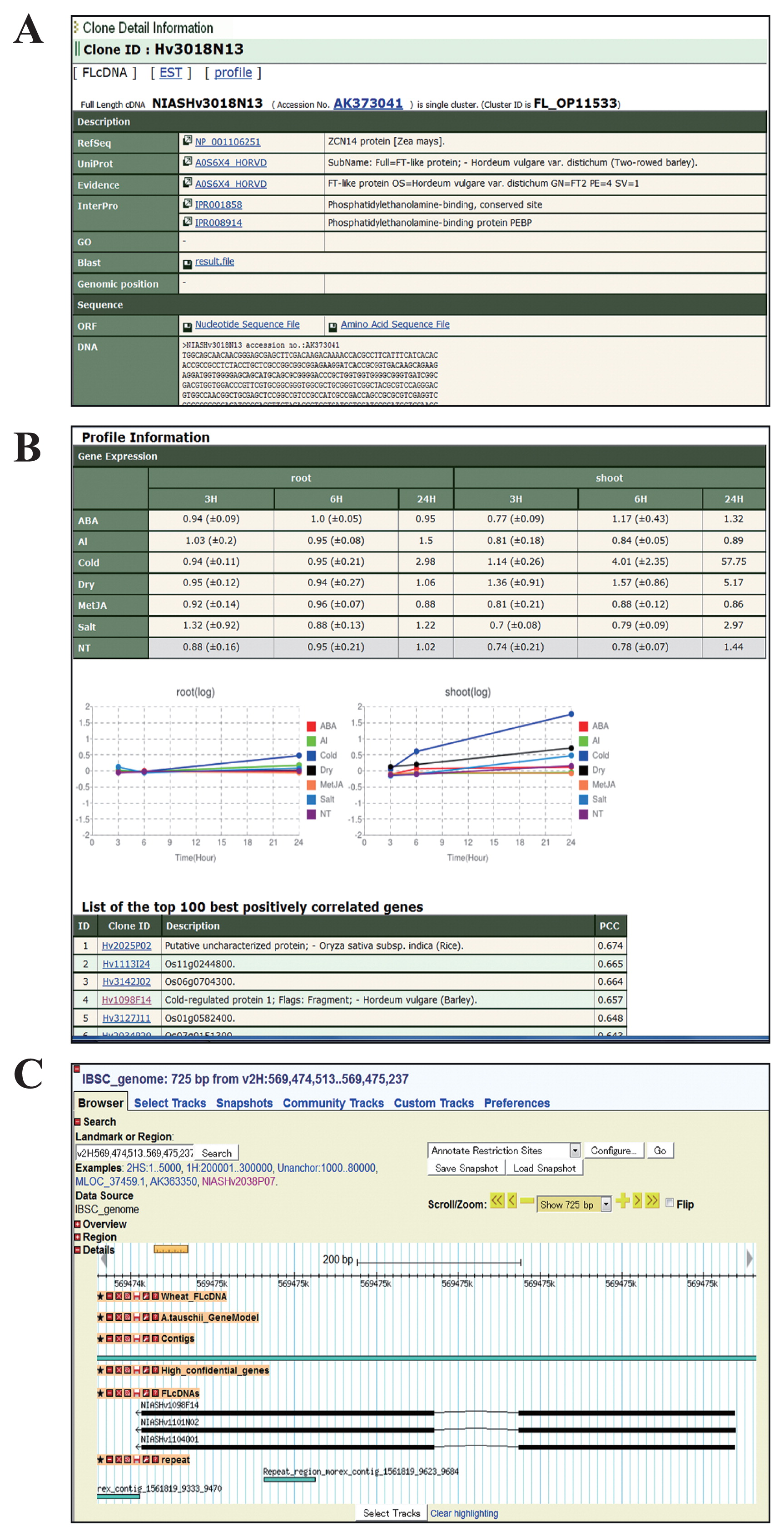
Information for construction and sequencing of 12 ‘Haruna Nijo’ cDNA libraries is available on the top page (Project Summary) of bex-db (http://barleyflc.dna.affrc.go.jp/bexdb/pages/help/project_summary.html). FLcDNA data represent the completed nucleotide sequences analyzed in 4,999 clones (FLbaf) by Okayama University (http://www.shigen.nig.ac.jp/barley/) and 24,783 clones (NIASHv) by the National Institute of Agrobiological Sciences, respectively (Fig. 1) (Sato et al. 2009, Matsumoto et al. 2011). Clustering of the above FLcDNAs was performed as reported previously, which led to the identification of 4,543 clusters (FL_CL) and 18,117 singletons (FL_OP) in the current database (Matsumoto et al. 2011). To attach annotations to the above FLcDNA sequences, we predicted the open reading frames and gene functions by BLASTX search (Altschul et al. 1990) using the RefSeq database (NCBI Resource Coordinators 2013) and the UniProtKB database (Magrane and UniProt Consortium 2011). The InterProScan software (Mulder and Apweiler 2007) was also used to assign both InterPro domains (http://www.ebi.ac.uk/interpro/) and Gene Ontology annotations (http://www.geneontology.org/) to the FLcDNA sequences (Fig. 2A).
Additionally, we released the sequences of 309,117 new ESTs (DK584720–DK887267) derived from 167,596 cDNA clones to the bex-db at this study (Fig. 1). The above ESTs constituted 141,521 pairs of 5′-end and 3 ′-end sequences and 26,075 single sequences from either the 5′- or 3′-end of the clones. On the basis of their sequence similarity analyzed using the EST clustering programs of TGICL (http://compbio.dfci.harvard.edu/tgi/software/) and CAP3 (Huang and Madan 1999) or our in-house re-clustering program when necessary, we were able to construct 27,562 contigs (Hv-Contig) that could be visualized through the “Contig viewer” of our bex-db. Alignments and consensus sequences of these EST contigs are downloadable through the database. We also constructed 22,148 contig clusters (EST_CL), basing on the information of paired ends (http://barleyflc.dna.affrc.go.jp/bexdb/pages/help/clustering_method.html), to be displayed in a “Cluster viewer”. On the other hand, 3,380 EST sequences (EST_OP) are known to be present as singletons within the database. Library names and clone information for each EST were also included in the above viewers. In order to provide users with more information on the predicted functions of barley genes, moreover, a BLASTX search (Altschul et al. 1990) of all consensus sequences of EST contigs (Hv-contigs) against the RefSeq database (NCBI Resource Coordinators 2013) and the UniProtKB database (Magrane and UniProt Consortium 2011) was also conducted. This information can be easily accessed by a keyword search as described later.
Microarray data
A 60-mer oligonucleotide microarray was developed to characterize the gene expression levels in barely using different experimental samples and conditions (Nakamura et al. in preparation). The 4x44K customized microarray platform (Agilent Technologies) contains the probes designed from 36,632 barley FLcDNAs. Our bex-db provides the current results of expression profiles of barley genes obtained from the above microarray analysis using the experimental samples and conditions as follows: root and shoot treated with or without abscisic acid (ABA), jasmonic acid (JA), cold, drought, aluminum and salt stress by 3 h, 6 h and 24 h (Fig. 2B). For comparison of the expression profiles among different genes, we calculated Pearson’s correlation coefficient of clone pairs on the array using their relative expression levels. A list of the top 100 genes (probes) with positively or negatively correlated expression patterns to each other was created for each microarray dataset.
Mapping of FLcDNAs onto the barley genome
A genome browser to display the annotation data of barley genes reported by IBSC has already been developed within a database, EnsemblPlants (http://plants.ensembl.org/Hordeum_vulgare/Info/Index/). This database, however, does not contain any information relating to the sequence analysis of barley FLcDNAs. To provide additional information on the barley genome, such as physical positions and structures of expressed genes or composition and chromosomal distribution of repetitive sequences, we thereafter reconstructed the genome viewer with GBrowse 2.54 software (Stein et al. 2002) using the genomic sequence from the barley variety ‘Morex’ published by IBSC (2012). The IBSC sequence of the barley genome currently consists of 2,670,738 assembled contigs with 1.9 Gb nucleotides. These sequence contigs were then assigned to the short (HS) and long (HL) arms of all seven barley chromosomes except for chromosome 1H, according to the orders indicated in EnsemblPlants. Some contigs remained unassigned because their chromosomal positions could not be determined (vHS, vHL and unanchored contigs). After masking for repeats by the software tool CENSOR (http://www.girinst.org/downloads/software/censor/) with mipsREdat_9.0p_Poaceae_TEs as described previously (Nussbaumer et al. 2013), we were able to map 11,758 barley FLcDNA sequences onto the barley chromosomes through homology searches using BLASTN (>95% identity and >90% coverage) (Altschul et al. 1990) and EST2genome (Mott 1997) in the present study (Fig. 2C).
Besides the above barley FLcDNAs, we were also able to map tentatively 50,554 sequences of the 83,382 wheat mRNAs deposited in DDBJ/EMBL/GenBank and 23,017 of the 43,150 gene models predicted in Aegilops tauschii (Jia et al. 2013) on the above barley genomic sequence (identity >70% and a coverage >50%). These important results should provide additional resources for future comparative and functional genomics studies within the Triticeae.
Search functions
The database contains convenient tools for users to access all data contents through a keyword search, sequence homology search and expression profile search from the top page, as described below (Fig. 1). All of the data are integrated by clone IDs, moreover, enabling users to see different results obtained from our experiments and analysis together with associated information through a cross-link function.
Keyword search
A keyword search can be used directly to obtain basic information about ESTs and FLcDNAs, including their sequences and functional annotations based on UniProtKB and RefSeq databases. Keywords such as accession numbers, clone IDs, gene names, or any word associated with the gene function can be used for the search. As a result, the top 50 BLAST hits will be shown in a list. In this case, users can choose to display the results obtained from all species or from a limited number of species and even a single species based on their interests. The above results can also be sorted easily by the BLAST hit score or its E value.
Homology search
A BLAST search engine (Altschul et al. 1990) is provided to serve as the tool for comparative analysis of the gene sequences in this study. A search can be performed using the barley FLcDNA or EST sequences from the ‘Haruna Nijo’ variety and the whole genome sequence of the ‘Morex’ variety. Depending on the query content, users can choose BLASTN or TBLASTX for analysis of the nucleotide sequences and TBLASTN for analysis of the amino acid sequences. These searches generate a list of homologous sequences matched by the query, including the alignments. For each hit sequence, a link to the basic clone information or the genome browser is also provided for use.
Expression profile search
A search for the expression profiles of barley genes can be effectively initiated by specifying the samples (root/shoot) of experimental materials, the experimental conditions and the time points of any treatment (3 h, 6 h, 24 h) used for microarray analysis and by setting the fold change and cutoff of expression levels. Results derived from the above keyword searches are shown in tabular format, indicating the IDs of hit clones and the exact fold changes under a given condition together with other information such as the accession numbers and cluster IDs of FLcDNAs, the IDs of EST contigs as well as the functional annotation based on BLASTX analysis against UniProtKB and RefSeq databases. There exists a link for each clone ID to its expression profile showing the full dataset obtained from microarray analysis in both tabular and graph formats and a list of its co-expressed genes. The above search function can be expanded, furthermore, using the “add” button to create expression profiles across a number of various treatments and experimental conditions.
Genome browser
To view genomic information about barley, users can choose either a homology search of sequences or direct access to the genome browser (GBrowse) from the top page of our bex-db. For homology searches, the IBSC genome sequence serves as a database which links the hit contigs directly from the BLAST result to the genome browser. A keyword search against GBrowse can be performed using the accession numbers of the FLcDNAs of interest to view their physical locations on the barley genome. Additionally, the IBSC gene IDs and the transcript or gene IDs of other different species can also be applied for the keyword search for the same purpose. All of the FLcDNA sequences mapped on the barley genome inside the GBrowse have a link to their clone information. It should be noted that not all of the barley cDNA clones have been completely sequenced or used for microarray analysis to date. All of the cDNA clones, however, have information on EST sequences with contig and/or cluster IDs that should enable users to find other clones assembled within the same contigs or clusters to find data about FLcDNA sequences and gene expression profiles. To enrich the above information, further chromosomal mapping of all EST sequences must be conducted.
An example of verifying candidate genes or gene functions using bex-db
The FLOWERING LOCUS T (FT)-like genes are involved mainly in controlling flowering signals and are regulated by photoperiod and vernalization pathways in barley (Faure et al. 2007). A keyword search against our bex-db using ‘FT-like protein’ or a homology search using the HvFT2 protein sequence from UniProtKB (A0S6X4) led to one result for one FLcDNA clone Hv3018N13 (accession no. AK373041) (Fig. 2A). The expression profile indicates that this gene is strongly expressed under cold stress with more than 50-fold expression level in the shoots after 24 hrs of treatment (Fig. 2B). This result can also be obtained by searching the data on gene expression profiles under certain experimental conditions, such as cold stress, shoot, 24 hrs and >50-fold changes. Chromosomal mapping of the above FLcDNA sequence by BLASTN analysis against the IBSC genomic sequence showed clearly that this gene was located within the Morex_contig_1558556 of barley chromosome 3HS, consistent with a previous report (Faure et al. 2007). By comparing the expression profiles among different genes, furthermore, a barley FLcDNA clone, NIASHv1098F14 (accession no. AK359508), which was predicted to encode the gene for cold-regulated protein 1, was found to be listed among the top 10 best positively correlated genes according to Pearson’s correlation coefficient calculation. The above results thereafter clearly demonstrated that the bex-db constructed in the present study could be a useful tool not only for comparative genomics among cereal crops, but also for the future identification of gene functions in barley.
Clone order
The barley FLcDNAs clones are available for distribution by the National Institute of Agrobiological Sciences by sending a request to the NIAS DNA Bank (http://www.dna.affrc.go.jp/distribution/).
Acknowledgements
We would like to thank Dr. B. Antonio for critical reading of this manuscript. This work was supported by grants from the Ministry of Agriculture, Forestry and Fisheries of Japan (Genomics for Agricultural Innovation TRG-1008 and GIR-1001, Genomics-based Technology for Agricultural Improvement TRS-1001).
Literature Cited
- Altschul, S.F., W. Gish, W. Miller, E.W. Myers and D.J. Lipman (1990) Basic local alignment search tool. J. Mol. Biol. 215: 403–410.
- Cockram, J., T. Thiel, B. Steuernagel, N. Stein, S. Taudien, P.C. Bailey and D.M. O’Sullivan (2012) Genome dynamics explain the evolution of flowering time CCT domain gene families in the Poaceae. PLoS One 7: e45307.
- Faure, S., J. Higgins, A. Turner and D.A. Laurie (2007) The FLOWERING LOCUS T-like gene family in barley (Hordeum vulugare). Genetics 176: 599–609.
- Huang, X. and A. Madan (1999) CAP3: A DNA sequence assembly program. Genome Res. 9: 868–877.
- Jia, J., S. Zhao, X. Kong, Y. Li, G. Zhao, W. He, R. Appels, M. Pfeifer, Y. Tao, X. Zhang et al. (2013) Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496: 91–95.
- Kikuchi, R., H. Kawahigashi, M. Oshima, T. Ando and H. Handa (2012) The differential expression of HvCO9, a member of the CONSTANS-like gene family, contributes to the control of flowering under short-day conditions in barley. J. Exp. Bot. 63: 773–784.
- Magrane, M. and UniProt Consortium (2011) UniProt Knowledgebase: a hub of integrated protein data. Database 2011: bar009.
- Matsumoto, T., T. Tanaka, H. Sakai, N. Amano, H. Kanamori, K. Kurita, A. Kikuta, K. Kamiya, M. Yamamoto, H. Ikawa et al. (2011) Comprehensive sequence analysis of 24,783 barley full-length cDNAs derived from 12 clone libraries. Plant Physiol. 156: 20–28.
- Mott, R. (1997) EST_GENOME: a program to align spliced DNA sequences to unspliced genomic DNA. Comput. Appl. Biosci. 13: 477–478.
- Mulder, N. and R. Apweiler (2007) InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol. Biol. 396: 59–70.
- NCBI Resource Coordinators (2013) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 41: D8–D20.
- Nussbaumer, T., M.M. Martis, S.K. Roessner, M. Pfeifer, K.C. Bader, S. Sharma, H. Gundlach and M. Spannagl (2013) MIPS PlantsDB: a database framework for comparative plant genome research. Nucleic Acids Res. 41: D1144–D1151.
- Sato, K., T. Shin-I, M. Seki, K. Shinozaki, H. Yoshida, K. Takeda, Y. Yamazaki, M. Conte and Y. Kohara (2009) Development of 5006 full-length CDNAs in barley: a tool for accessing cereal genomics resources. DNA Res. 16: 81–89.
- Stein, L.D., C. Mungall, S. Shu, M. Caudy, M. Mangone, A. Day, E. Nickerson, J.E. Stajich, T.W. Harris, A. Arva et al. (2002) The generic genome browser: a building block for a model organism system database. Genome Res. 12: 1599–1610.
- The International Barley Genome Sequencing Consortium (2012) A physical, genetic and functional sequence assembly of the barley genome. Nature 491: 711–716.