Abstract
Soybean (Glycine max) has a paleopolyploid genome, and many re-sequencing experiments to characterize soybean genotypes have been conducted using next-generation sequencing platforms. The accumulation of information about single nucleotide polymorphisms (SNPs) throughout the soybean genome has accelerated identification of genomic regions related to agronomically important traits through association studies. However, although many efficient mapping techniques that use next-generation sequencing are available, the number of practical approaches to identify genes/loci is still limited. In this study, we used a combination of restriction site–associated DNA sequencing (RAD-seq) and bulk segregant analysis (BSA) to identify quantitative trait locus (QTLs) for flowering time in a segregating population derived from a cross between Japanese soybean cultivars. Despite the homogeneous genetic background of the parents, over 7000 SNPs were identified and can be used to detect QTLs by RAD-seq BSA analysis. By comparing genotype frequency between early and late-flowering bulks from the F3 segregating population, we identified a QTL on Gm10, which corresponds to the previously identified E2 locus, and a QTL on Gm04, which is close to the E8 locus. Out of these SNPs, more than 2000 were easily converted to conventional DNA markers. Our approach would improve the efficiency of genetic mapping.
Introduction
Soybean (Glycine max) is a crop important for oil production, for human consumption, and for livestock feeds. The production of soybean is steadily increasing (FAO; http://faostat.fao.org/). To meet the increasing demand for food, plant breeding techniques need to be improved. Efficient use of genome information is necessary to accelerate soybean breeding. Soybean genome sequence is freely available since 2008 in Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html). The estimated soybean genome size is 1.1 Gbp (including the assembled sequence of 969.6 Mbp); over 46000 genes have been predicted and 75% of them are present in multiple copies (Schmutz et al. 2010). The genomes of wild soybean (Glycine soja, Kim et al. 2010) and the elite Japanese soybean cultivar Enrei (Katayose et al. 2012, Shimomura et al. 2015) have also been sequenced.
Owing to the advances in sequencing technology, the number of large-scale re-sequencing studies, such as re-sequencing of 302 wild, landrace, and cultivated soybeans (Zhou et al. 2015), has also increased. Information on genomic SNPs from a large number of lines of different genetic backgrounds enables whole-genome association studies, revealing genomic regions associated with soybean domestication. Several candidate loci controlling agronomically important quantitative traits such as oil content and yield-related characters have been identified (Zhou et al. 2015). The validations of these quantitative trait loci (QTLs), including their availability for soybean breeding, are needed. However, the confirmation of minor QTLs in relevant mapping populations showing segregation of genes related to these QTLs and the construction of a genome-wide genetic map with traditional DNA markers, such as simple sequence repeat (SSR) markers, are still time-consuming and laborious. To facilitate mapping, several SNP-based genotyping platforms were developed. High-throughput genotyping systems, such as the GoldenGate assay (Illumina, Inc., San Diego, CA, USA), Infinium BeadChips (Illumina, Inc.), and the MassARRAY system (Sequenom, Inc., San Diego, CA, USA) were used to evaluate the genetic diversity of soybean germplasm (Kaga et al. 2012, Song et al. 2013) and to construct a high-density linkage map containing 5500 DNA markers (Hyten et al. 2010). These genotyping systems could provide high-throughput mapping techniques for soybean breeding programs; however, they require considerable initial investment in equipment. Hence, cost-effective mapping methods that do not need any specific equipment would be valuable for identification of agronomically important QTLs. Moreover, developing tightly linked DNA markers for target QTLs could accelerate marker-assisted selection.
Next-generation sequencing (NGS) platforms allow obtaining large numbers of short read sequences within a short period. Identification of rice genes, loci, and major QTLs related to agronomic traits by using whole-genome re-sequencing has been reported by Abe et al. (2012), who used a method termed Mut-seq, and by Takagi et al. (2013), who used QTL-seq. Both methods rely on bulk segregant analysis (BSA, Quarrie et al. 1999) with re-sequencing of bulked DNAs from several individuals selected from a segregating population on the basis of their phenotypes. As an alternative to whole-genome sequencing of bulked DNAs, a method to reduce genome complexity by using a restriction enzyme was developed and used to identify over 13,000 polymorphic sites in a target genome (Baird et al. 2008). In this method, Sequences adjacent to restriction sites are concentrated in a DNA library; this approach reduces the number of genomic regions to be sequenced and increases the number of reads per DNA fragment. Restriction site–associated DNA sequencing (RAD-seq) with massively parallel sequencing platforms is widely used for many species. A restriction enzyme change or using a combination of restriction enzymes increases the flexibility of this method (Poland et al. 2012). This method is applicable for mapping of qualitative loci using BSA of F2 populations (Baird et al. 2008).
To apply these techniques to soybean, we need to consider genome size, nucleotide composition, and polyploidy level, and to choose a suitable restriction enzyme and the range of DNA fragment sizes by using in silico simulation. Because the soybean genome is paleopolyploid and the proportion of paralogous genes is high (Schmutz et al. 2010), some difficulties in precise mapping of short reads and SNP calling are expected. To directly apply the QTL-seq method developed in rice to QTL mapping in soybean, a larger number of reads would be necessary because the size of the soybean genome is larger than that of the rice genome, and the large number of SNPs obtained from whole-genome sequencing would hamper finding the location of the target QTL. On the other hand, RAD-seq analysis also has problems, especially low diversity of a base close to the sequencing primer, which reduces the quality of sequencing data, because digested DNA fragments would have common nucleotide sequences corresponding to the restriction sites (Mitra et al. 2015).
In this study, we used RAD-seq and BSA to identify QTLs related to flowering time in a soybean mapping population derived from a cross between two Japanese soybean cultivars (G. max). We ligated the adapter-containing index sequence before DNA bulking and increased the number of restriction enzymes to avoid the low-diversity problem. Procedures for developing DNA markers tightly linked to target QTLs reported here would extend the utility of RAD-seq for mapping experiments. Unlike the web-based primer design tools such as “derived cleaved amplified polymorphic sequence (dCAPS) Finder 2.0” (Neff et al. 2002), our script can deal with a massive number of SNPs at the same time and outputs a list of dCAPS markers with different restriction enzymes from which the user can choose. These improvements could facilitate developing DNA markers for breeding programs in soybean and other species.
Materials and Methods
Plant materials and phenotypic analysis
Japanese cultivars ‘Fukuyutaka’ (hereafter Fuku) and ‘Toyoshirome’ (Toyo) (both G. max), bred at the Kyushu Okinawa Agricultural Research Center, National Agriculture and Food Research Organization, with the aim of high-yielding cultivars in the southern part of Japan, especially the Kyushu area. We crossed these cultivars and obtained an F3 mapping population containing 155 plants derived from each of the F2 individuals by single-seed descent. The date of first flowering (R1), flowering time of the top raceme (R2), and harvest date (between R7 to R8) were recorded for each F3 plant and parental lines according to Fehr’s measurement criteria (Fehr et al. 1971). Seeds of F2 plants were sown in the field of Saga University (33°14N 130°17E) on 21 July 2015 and plants were grown in accordance with standard cultivation practice under natural day length.
Construction of an NGS library
Total DNA was extracted from a young leaflet of each F3 plant and parents (with two replications) with the CTAB method (Murray and Thompson 1980). Among the F3 population, we chose 12 early flowering plants and 12 late-flowering plants. DNA (500 ng) was digested with CivQI (8 units; New England Biolabs (NEB) Japan Inc., Tokyo, Japan) at 25°C for 3 h, and the enzyme was inactivated by heating at 65°C for 20 min. DNA was precipitated with ethanol, and DNA digestion was verified by agarose gel electrophoresis. Half of the DNA digested with CivQI (250 ng) was further digested with a mixture containing both PstI and MspI (8 units each, NEB) at 37°C for 3 h, followed by enzyme inactivation at 65°C for 20 min. Of the three pre-annealed adapters, one contained a PstI overhang and an index sequence (capitalized): 5′-cacgacgctcttccgatct CATCACAAGtgca-3′ and 5′-CTTGTGATGagatcggaagagcgtcgtg-3′. The other two were Y-adapters for MspI (5′-CGAGATCGGAAGAGCGGGGACTTTAAGC-3′ and 5′-GATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT-3′) and CivQI (5′-TAAGATCGGAAGAGCGGGGACTTTAAGC-3′ and 5′-GATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT-3′). Adapters were prepared as described in Poland et al. (2012); the adapter sequences were modified according to the restriction site sequence. Index sequences corresponding to 28 DNA samples are summarized in Table 1. The adapters were ligated to corresponding digested DNA with T4 DNA ligase (350 units; Takara, Otus, Japan) at 22°C overnight. After ligation, samples were combined, purified on a silica spin column and separated by electrophoresis on a 2% agarose-L gel (Nippon Gene, Tokyo, Japan). The region containing 400–500 bp DNA fragments was excised, DNA was purified on a silica column and an aliquot (12 μl out of 50 μl) was used as a template for PCR with the primers P1 (5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCT-3′) and P2 (5′-CAAGCAGGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAA-3′). The PCR mixture (final volume of PCR mixture was 120 μl) also contained PCR buffer, dNTPs and PrimeSTAR GXL polymerase (Takara) according to the manufacturer’s instructions. PCR conditions were 98°C for 2 min, 12 cycles of 98°C for 10 s, 60°C for 15 s, and 68°C for 30 s, with 5 min final extension. PCR products were separated on a 1% agarose gel and the region containing 400–500 bp DNA was excised from the gel to eliminate a short-DNA fraction probably containing primer dimers and unspecific PCR amplification products; DNA was purified as above. The generated NGS library (500 ng DNA) was analyzed by BGI (Shanghai, China) on Hiseq 4000 (Illumina, Inc.), in 2 × 100 bp paired-end mode. Sequence data (approximately 30 Gbp) were classified according to index sequences mentioned above and were used for further analysis.
Table 1
Summary of NGS data obtained from each file separated with an indexed sequence
| ID |
Bulk or cultivar |
Indexed sequence |
Total reads (M)a |
Mapped pairs (M) |
Ratio of mapped sequences |
Average insert size (bp) |
Standard deviation of insert size |
Number of loci (K)a |
Average depth |
Median of depth |
Mode of depth |
Estimated genome coverage |
| A01 |
Early |
TGACGCCA |
9.2 |
6.5 |
70.5% |
336.2 |
44.8 |
113.1 |
57.1 |
1 |
6 |
2.3% |
| A02 |
Early |
GGCTTA |
11.8 |
8.1 |
68.4% |
337.1 |
44.9 |
123.3 |
65.7 |
1 |
7 |
2.5% |
| A03 |
Late |
CTAAGCA |
9.7 |
6.9 |
71.3% |
336.6 |
44.8 |
120.1 |
57.7 |
1 |
6 |
2.4% |
| A04 |
Fukuyutaka |
GCCTACCT |
9.7 |
6.6 |
68.5% |
336.5 |
44.8 |
103.5 |
64.2 |
1 |
6 |
2.1% |
| B01 |
Early |
CAGATA |
8.6 |
6.0 |
69.4% |
335.4 |
44.9 |
120.8 |
49.7 |
1 |
6 |
2.4% |
| B02 |
Early |
AACGCACATT |
10.9 |
7.7 |
70.6% |
334.3 |
44.8 |
121.4 |
63.6 |
1 |
6 |
2.4% |
| B03 |
Late |
ATTAT |
12.3 |
8.4 |
68.2% |
333.4 |
45.1 |
125.5 |
66.6 |
1 |
7 |
2.5% |
| B04 |
Fukuyutaka |
CACCA |
9.5 |
6.6 |
68.9% |
338.4 |
44.7 |
108.5 |
60.5 |
1 |
6 |
2.2% |
| C01 |
Early |
GAAGTG |
8.9 |
6.5 |
72.5% |
337.2 |
44.8 |
124.9 |
51.6 |
1 |
6 |
2.5% |
| C02 |
Early |
GAGCGACAT |
11.7 |
8.2 |
69.7% |
335.7 |
44.7 |
121.4 |
67.3 |
1 |
6 |
2.4% |
| C03 |
Late |
GCGCTCA |
11.0 |
7.6 |
69.1% |
336.6 |
44.6 |
116.7 |
65.1 |
1 |
6 |
2.3% |
| C04 |
Toyoshirome |
AATTAG |
10.5 |
7.5 |
71.5% |
336.6 |
44.9 |
108.0 |
69.8 |
1 |
7 |
2.2% |
| D01 |
Early |
TAGCGGAT |
10.9 |
7.6 |
69.8% |
336.0 |
44.7 |
126.2 |
60.1 |
1 |
6 |
2.5% |
| D02 |
Early |
CCTTGCCATT |
8.2 |
5.8 |
70.5% |
334.5 |
44.8 |
119.3 |
48.2 |
1 |
6 |
2.4% |
| D03 |
Late |
ACTGCGAT |
12.8 |
8.4 |
65.4% |
335.7 |
44.8 |
124.6 |
67.1 |
1 |
7 |
2.5% |
| D04 |
Toyoshirome |
GGAACGA |
11.1 |
8.0 |
71.9% |
338.0 |
44.7 |
104.3 |
76.3 |
1 |
6 |
2.1% |
| E01 |
Early |
TATTCGCAT |
9.5 |
6.7 |
69.9% |
335.7 |
44.8 |
117.8 |
56.6 |
1 |
6 |
2.4% |
| E02 |
Late |
GGTATA |
9.4 |
6.6 |
69.9% |
334.9 |
45.0 |
113.0 |
58.4 |
1 |
6 |
2.3% |
| E03 |
Late |
TTCGTT |
10.3 |
6.6 |
64.4% |
336.3 |
44.8 |
117.9 |
56.3 |
1 |
6 |
2.4% |
| F01 |
Early |
ATAGAT |
8.5 |
6.0 |
70.3% |
334.2 |
45.0 |
121.2 |
49.5 |
1 |
6 |
2.4% |
| F02 |
Late |
TCTTGG |
10.0 |
7.1 |
71.0% |
338.3 |
44.7 |
119.5 |
59.2 |
1 |
6 |
2.4% |
| F03 |
Late |
ATATAA |
12.0 |
8.6 |
71.6% |
330.7 |
45.0 |
115.6 |
74.6 |
1 |
7 |
2.3% |
| G01 |
Early |
CCGAACA |
9.1 |
6.4 |
70.3% |
336.8 |
44.7 |
125.5 |
51.0 |
1 |
6 |
2.5% |
| G02 |
Late |
GGTGT |
16.1 |
10.9 |
67.4% |
338.5 |
44.8 |
121.5 |
89.5 |
1 |
6 |
2.4% |
| G03 |
Late |
TGGCAACAGA |
11.2 |
7.7 |
69.0% |
334.3 |
44.7 |
112.6 |
68.5 |
1 |
6 |
2.3% |
| H01 |
Early |
GGAAGACAT |
8.7 |
6.2 |
70.6% |
335.3 |
44.8 |
121.9 |
50.6 |
1 |
6 |
2.4% |
| H02 |
Late |
GGATA |
10.1 |
7.2 |
71.7% |
335.8 |
44.9 |
118.4 |
61.1 |
1 |
6 |
2.4% |
| H03 |
Late |
CTCGTCG |
4.5 |
3.0 |
66.5% |
337.1 |
44.7 |
89.7 |
33.1 |
1 |
4 |
1.8% |
| ALL |
|
|
286.40 |
199.14 |
69.5% |
N. C.b |
N. C. |
284.4 |
677.6 |
1 |
13 |
5.7% |
a M, million; K, thousand.
The data mining steps and custom Perl scripts for soybean genome simulation, for NGS data analysis, and for designing primers for dCAPS markers (Neff et al. 1998) are summarized in Supplemental Fig. 1 and Supplemental Table 1. Briefly, to choose the suitable combinations of restriction enzymes, degree of the reduction of the target genome region for sequencing was calculated as the ratio between total read length (190 bp per fragment) of all fragments included in a specific size range and soybean genome size (approx. 960 Mbp). We mapped all reads on the soybean reference genome sequence (Gmax_189_hardmasked.fa) with Bowtie2 (Langmead and Salzberg 2012) with default parameters. Samtools and BCFtools were used to sort and index each file (SAM/BAM file), and the function “mpileup” was used to make variant call format (VCF) files by combining multiple SAM files. We made a VCF file containing all sequence data including 28 indexed samples. To eliminate the SNPs between the reference sequence (‘William 82’) and Japanese soybeans (Fuku and Toyo), we selected SNP/Indel (hereafter referred to as SNPs) sites between Fuku and Toyo with a genotype ratio (number of reads of the reference or alternative allele divided by the total number of reads at a locus) of 0.25–0.75 for reference and alternative alleles with read coverage >100×. We listed the positions of these SNPs to generate a candidate SNP list. Then we made six types of VCF files for all files combined, the early-flowering group, late-flowering group, each parental line, and mixed parental lines on the basis of this list of SNP sites. We selected SNPs with read coverage >24× (at least two reads from each of 12 individuals) in each early or late bulk. We identified skewed genotype distributions as follows:
|
Index score
=
|
log
10
R
a
t
i
o
o
f
r
e
f
e
r
e
n
c
e
a
l
l
e
l
e
i
n
e
a
r
l
y
b
u
l
k
+
0.001
R
a
t
i
o
o
f
r
e
f
e
r
e
n
c
e
a
l
l
e
l
e
i
n
l
a
t
e
b
u
l
k
+
0.001
| |
We added 0.001 to all scores to avoid division by zero; the maximum index score is expected to be 3. In addition, we calculated the threshold index score for the significance level of 0.01 with a permutation test with 1000 replications as follows. First, VCF files were made separately based on the index sequences of 24 of 28 samples (except for parental sequence data). These files were randomly combined into two groups (12 files each). The index scores were calculated for all loci and recorded. This procedure was then performed a total of 1000 times to obtain the threshold values at 1% significance levels. This calculation was performed with homemade functions running in R software (R Development Core Team 2008).
Confident SNPs between parents were selected from parental VCF files again under more stringent conditions that one parent showed a genotype ratio for reference or alternative alleles of >0.95 and the other parent showed <0.05, indicating homozygosity of each parent. The VCF file re-made based on the list of confident SNPs from all sequencing data was used as an input file for designing dCAPS primers. Custom Perl scripts were written to automatically perform the following four steps: 1) retrieving the sequence that included the SNP site from the Gmax_189.fa sequence data; 2) judging whether this sequence could be available for dCAPS with a particular list(s) of restriction enzymes (example file is shown in supplemental data); 3) designing primers to amplify the target SNP with primer3 (Untergasser et al. 2012); and 4) counting number of loci that identical to primer sequence with blast+ program (Camacho et al. 2009) to avoid PCR amplification of multiple loci in the genome. The final output file contained the following: information about chromosome and position of each locus, primer sequences, the name of the restriction enzyme, and the number of loci in the soybean genome showing high similarity to the primer sequences. Inexpensive restriction enzymes were chosen to detect dCAPS; the list of these 17 enzymes is provided in supplemental data. To confirm whether the designed primers can correctly amplify the target loci and detect polymorphism between the parental lines, we used 51 primer pairs selected by these custom Perl scripts to construct a linkage map of chromosome 10 (Gm10) with the F3 population using the Ant map program (Iwata and Ninomiya 2006) with the default parameters.
DNA marker analysis
PCR mixtures contained gDNA (30–50 ng), 1 pmol each dCAPS primer, 0.1 U of homemade recombinant Taq polymerase, and the appropriate PCR buffer with 10 μl reaction volume. Primer sequences are listed in Supplemental Table 2. PCR conditions were 95°C for 5 min, 40 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s, with a final extension at 72°C for 5 min. PCR products were digested for 3 h with 1 U of a restriction enzyme according to the manufacturers’ instructions, separated by 12% polyacrylamide gel electrophoresis, stained with EtBr and visualized using a UV transilluminator. These dCAPS primers and PCR conditions were used to detect functional SNPs underlying recessive allele of the E2 gene caused by premature stop codon (Tsubokura et al. 2014). Primer sequences for an SSR marker used to confirm the presence of additional QTLs are also listed in Supplemental Table 2. PCR was performed as above; PCR product digestion was omitted.
Statistical analysis
We performed QTL analysis using the linkage map of Gm10 with the R/qtl package (Broman et al. 2003) and input data with default parameters for F2 population. Composite interval mapping method (Zeng 1993) with the number of covariates 1 and window size of 10 cM was used to display the logarithm of the odds (LOD) peak curve for the E2 locus. Additive and dominance effects, and phenotypic variance explained by the E2 gene and other candidate QTLs were estimated by linear regression analysis with the genotypes of E2 and other DNA markers tightly linked to the target QTL(s) and phenotypic values of each trait in individual plants. All statistical tests were performed in R (ver. 3.3.1) software (R Development Core Team 2008).
Results
Phenotypic distribution of flowering time in the F3 population and its genetic heritability
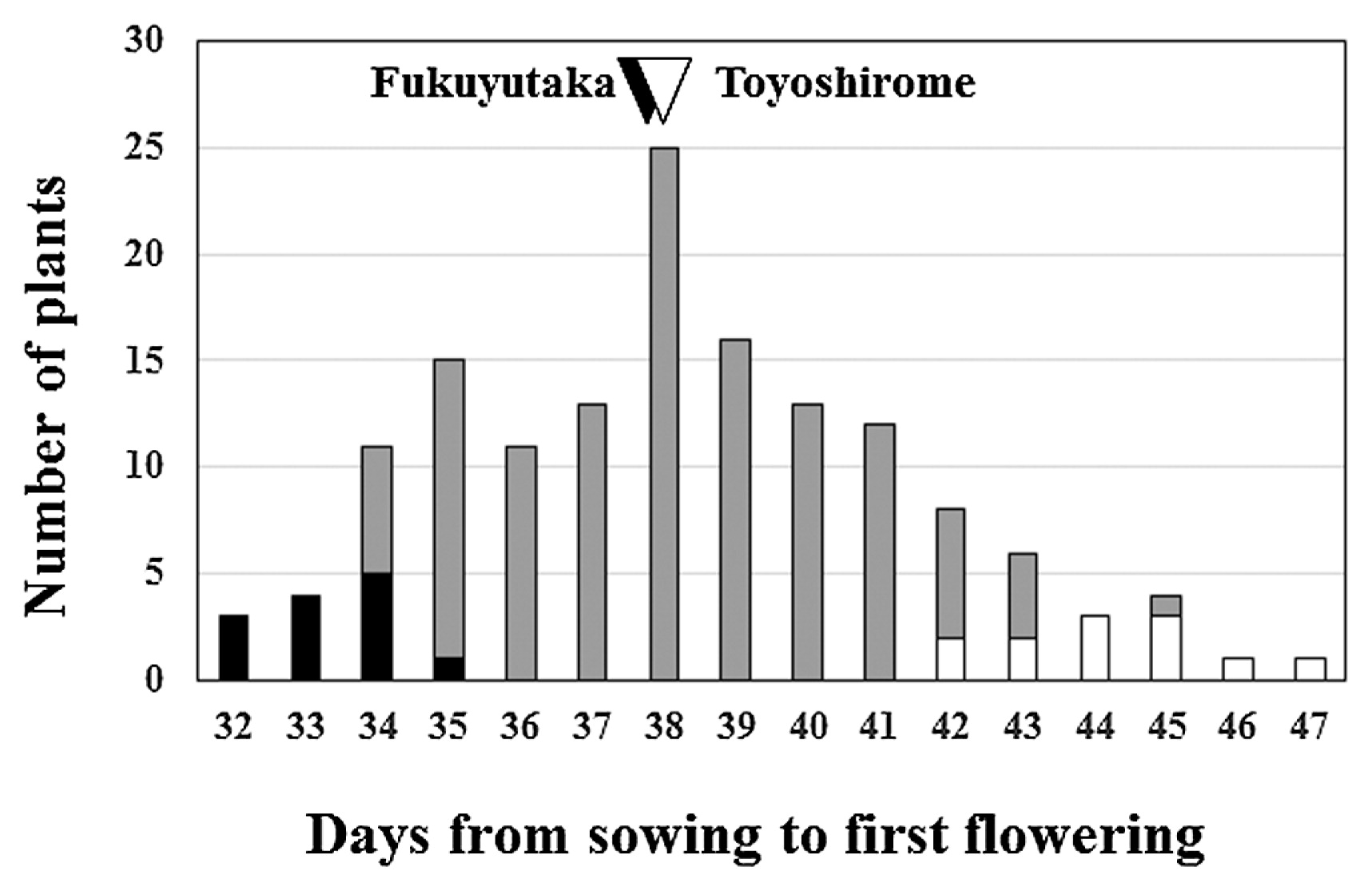
The difference between the flowering times of the parents, Toyo (average flowering time 37.7 ± 1.3 days) and Fuku (37.5 ± 1.5 days), was not significant (P > 0.05), whereas the range of flowering times observed in the F3 population (hereafter designated as TFF3) was extended in both directions (Fig. 1), indicating that some genes controlling flowering time were segregated in this population. Broad-sense heritability estimated by comparing the variance of the flowering time between TFF3 and parents was 0.79. Correlation between days to R1 and R2 was 0.92 and that between days to R1 and days to harvest was 0.67. High correlation values indicated that the genes controlling R1 phenotype also affected R2 and days to harvest. We made two TFF3 bulks (12 plants each), early flowering (average flowering time, 33.3 ± 1.0 days) and late flowering (44.2 ± 1.5 days), and used NGS analysis to identify the genomic regions associated with the difference between flowering phenotypes in the two bulks.
To select suitable combinations of restriction enzymes for DNA library construction, we analyzed in silico the pattern and size distribution of fragments obtained by restriction enzyme digestion of the soybean reference genome sequence (Gmax_189.fa). Poland et al. (2012) used a combination of PstI (for the index) and MspI (for the Y-adapter); however, the use of a single restriction enzyme for the Y-adapter can cause low diversity of the recognition site sequence in pair-end sequencing. We chose not only MspI (recognition site, CCGG) but also CviQI (GTAC) for Y-adapters. CviQI does not digest adapters or indexes, produces cohesive ends, shares no sequence identity with the MspI recognition site, and does not drastically increase the degree of reduction of the target region. The result of in silico analysis showed that PstI, MspI, and CviQI were suitable for sequencing and 0.38% of the soybean genome would be covered with 100-bp paired-end sequences if 400–500-bp DNA fragments are selected after digestion with these enzymes (Supplemental Table 3). Extending the size range to 100–500 bp increased genome coverage by sequencing to 1.77%. We expected that the reduction of complexity of the soybean genome under the above conditions would be sufficient to cover each locus with many reads, and prepared a DNA library accordingly.
Evaluation of NGS data
Twenty-eight NGS data files, which were grouped according to index sequences included in the PstI adapter, contained 4.5–12.3 million reads (average, 10.2 ± 2 million). Information related to the NGS data is summarized in Table 1. Approximately 70% of the read pairs were successfully mapped on the reference soybean genome (Gmax189_HM; hard-masked sequence). The percentage of mapped reads was higher than 30–40% in our previous RAD-seq experiment that used HaeIII (data not shown). Hence, the combination of restriction enzymes is an important factor affecting the percentage of mapped reads. The positions of paired mapped reads provided information about the size distribution of DNA fragments and sequenced genome regions present in the library. The length of DNA fragments extracted from the gels was 400–500 bp. Taking into account 70-bp adapters, the distribution of DNA sizes (335 ± 45 bp) was in good agreement with the expected distribution. The distribution of the number of independent loci separately mapped using each NGS file ranged from 89.7 to 130.0K loci (hereafter, K means ‘thousand’). The total number of independent mapped loci calculated from all NGS files was 284.4K. The number of loci with a single read from at least 18 out of 28 individuals (64.3%) was 91K. This score indicated that the short reads for many loci originated from different indexed samples. The read coverage for each locus from a single individual was however low; the mode of the distribution was 1 for all indexes and the median was 6. The total coverage of the soybean genome in this library was expected to be 5.6% (total read length [total number of loci, 284.4K, ×190 bp] divided by the size of the soybean genome [960 Mbp]). This value was higher than that in simulation, indicating the likely presence of unexpected fragments in DNA samples. However, the number of mapped loci was sufficient to find the chromosomal region(s) with skewed genotype distribution between early and late bulks because reads in the bulked DNAs were expected to originate evenly from multiple individuals, although the number of reads from each individual was low.
Chromosome region showing skewed genotype ratio between early and late bulks
We obtained NGS data separately for 28 individuals. The SNPs were extracted from the VCF files. We prepared bulks containing all individuals (Bulk 1), early flowering plants (Bulk 2), late-flowering plants (Bulk 3), mixed parents (Bulk 4), Fuku (Bulk 5), and Toyo (Bulk 6). To increase the number of reads from parents, we prepared two sample replications for each parent. In Bulk 1, the number of SNPs between the reference genome (‘Williams 82’) and the parental lines was 120K. We extracted 10,813 SNPs between parents, Fuku and Toyo. This number indicated that the genetic similarity between parents was much higher than their similarity to ‘Williams 82’. The estimated polymorphic ratio of Fuku and Toyo was 0.47 nucleotides/kbp (calculated as the number of SNPs obtained from all indexes divided by the length of independent reads estimated from all indexes).
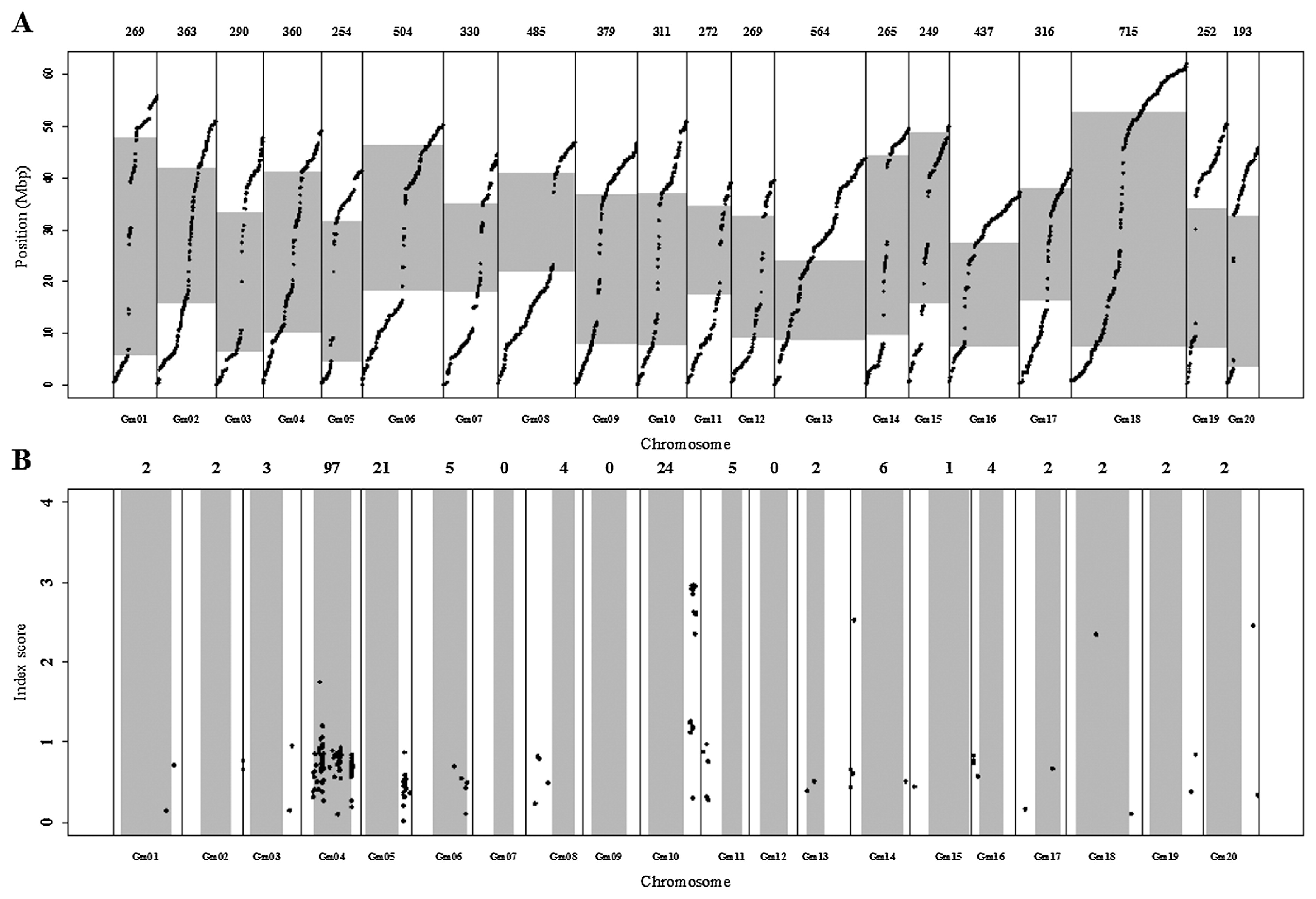
To identify the chromosome region(s) related to the difference between early and late flowering bulks, we used 7077 SNP loci that had >100× coverage in Bulk 1 and at least 24× coverage in at least Bulk 2 or Bulk 3. The physical positions of these SNPs were scattered evenly over the soybean genome (Fig. 2A), we considered the 7077 loci to be sufficient to identify the locus showing a skewed genotype ratio. We calculated the index score for each locus between Bulks 2 and 3 (Fig. 2B). A high index score value (a large difference in genotype frequency at a locus between two bulks) indicates that a QTL related to flowering time is located close to this locus. A Gm10 region showed the highest index score (2.96 for an SNP at 4,4506,176 bp), indicating the strongest bias between the two bulks. This skewed region expanded over 42.5–45.9 Mbp, where 17 of 20 SNPs had index scores >2.3. We also found two SNP clusters with index scores exceeding the threshold values calculated for each locus by permutation test. One cluster was located on Gm04 (highest index score of 1.75 for an SNP at 15,586,813 bp) and the other one on Gm05 (highest index score of 0.87 for an SNP at 36,142,493 bp). We considered these regions as candidate flowering time QTLs, but focused mainly on the locus on Gm10. We developed dCAPS markers linked to this QTL and determined its position more precisely using TFF3.
We developed custom Perl scripts to design primers for dCAPS. First, we selected 6872 high-confidence SNPs out of 10,853 SNPs to develop CAPS or dCAPS markers based on allele information obtained from Bulks 5 and 6 (parental genotypes). In case of only CAPS markers, only 470 SNPs were available to detect polymorphisms with 17 inexpensive restriction enzymes (the list of restriction enzymes is included in the Perl script files). In case of dCAPS, the number of markers increased to 2,163. When we screened SSR markers developed previously (Song et al. 2004), only 189 of 1152 markers (16.4%) showed polymorphism between the parents (Fuku and Toyo). We confirmed the reliability of the selected dCAPS markers by constructing a linkage map of soybean Gm10 with TFF3 and performed QTL analysis to estimate the precise location and genetic effects of the QTL. Clear polymorphic bands were observed in 46 out of 51 dCAPS markers (90.2%, Supplemental Table 2, Fig. 3A). We constructed a linkage map of Gm10 with 30 DNA markers (Fig. 3B). The genetic order of DNA markers matched well the order of their physical positions, and the relationship between genetic and physical positions on the linkage map matched a typical pattern of a relationship between a linkage map and a physical map, with a low ratio of recombination to physical distance in the pericentromeric region. We also identified a LOD score peak at 44.47–45.92 Mbp on Gm10 (Fig. 3B, 3C).
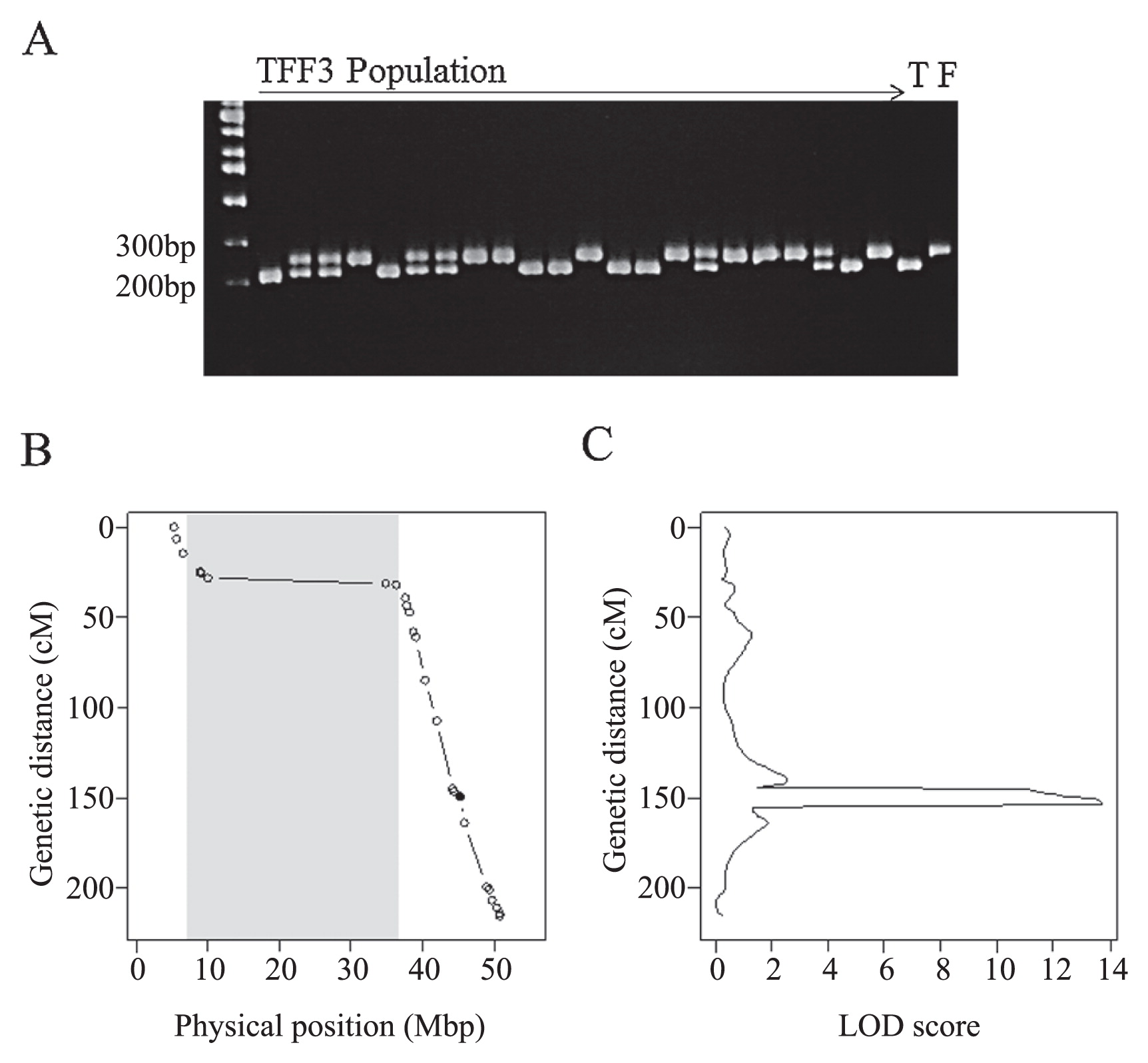
We examined the genes located in this region of Gm10 using the soybean genome dataset (Gmax_189) and found that the well-known soybean flowering gene E2 is located at 45.3 Mbp (Watanabe et al. 2011). Therefore, we analyzed the segregation of a functional SNP (FNP) causing a stop-codon mutation identified previously (Tsubokura et al. 2014). This FNP segregated in TFF3 and its position (149.3 cM) on the linkage map matched well the physical position of E2 (Fig. 3B, Supplemental Table 2). A LOD peak was also detected at 153 cM and the marker proximal to this peak was the FNP of E2 (Fig. 3C). Fuku had the functional late-flowering E2 allele, whereas Toyo had the early flowering e2 allele. This difference affected not only days to first flowering (R1) but also days to flowering of the top raceme (R2) and days to harvest (Table 2). We also used SSR and dCAPS markers to examine the presence of QTLs on Gm04 and Gm05. Linear regression analysis using the genotypes of these markers and flowering phenotypes showed significant association between phenotype and the genotype of the SSR marker AW277661, which is proximal to another soybean flowering gene, E8, located in the pericentromeric region of Gm04 (Watanabe et al. unpublished data). Fuku had an early flowering allele, and Toyo had a late flowering allele at this QTL (Table 2). Meanwhile, the genotypes of the dCAPS marker Gm05_36142849_TaqI showed no significant association with flowering phenotype (Table 2). The genetic effects of E2 and the QTL on Gm04 explained 41.0% of total phenotypic variance in flowering time in TFF3.
Table 2
Genetic effects estimated by linear regression analysis for candidate QTL loci
| Trait |
E2 |
AW277661(Gm04) |
Gm05_36142849_TaqI |
| Additive effecta |
Dominance effect |
Additive effect |
Dominance effect |
Additive effect |
Dominance effect |
PVEb |
| First flowering day from sowing (R1) |
−2.1c |
ns |
1.3 |
ns |
ns |
ns |
41.0% |
| Flowering day of the top raceme (R2) |
−3.2 |
ns |
1.5 |
ns |
ns |
ns |
54.3% |
| Harvest date (R7–R8) |
−3.4 |
ns |
2.1 |
ns |
ns |
ns |
32.9% |
a Genetic effects of the Toyoshirome allele compared to the Fukuyutaka allele.
b Phenotypic variance explained by the QTLs.
c Genetic effects significant at a level below 0.001 are shown; ns, not significant (
p ≥ 0.001).
Discussion
In this study, using Rad-seq BSA, we identified two QTLs segregating in a population derived from a cross between Japanese soybean cultivars. Both cultivars, Fuku and Toyo, are adapted to the Kyushu area and have similar flowering and maturity phenotypes and yield (Ohba 1980, 1985). Phylogenetic analysis using SNPs showed that these cultivars fall into the category of Japanese soybean cultivars. Fuku is included in the cluster J8 and Toyo in the cluster J1b (Kaga et al. 2012). This difference reflects the pedigree of these cultivars. Fuku was derived from a cross between the landraces ‘Okadaizu’ and ‘Shirodaizu-3’. Toyo was derived from a cross between the cultivars ‘Tosan-25’ and ‘Tamahomare’; both were bred in Nagano Prefecture, a mid-latitude region of Japan. Toyo and Fuku originated from different genotypes, but sequence diversity between them (0.47 nucleotides/kbp) is lower than sequence diversity (0.83 nucleotides/kbp) among 25 elite soybean cultivars developed in the United States before the 1980s (Hyten et al. 2006). The low number of polymorphic SSR markers between Fuku and Toyo also supports their similar genetic backgrounds. Despite nucleotide sequence similarity of these parental lines, we obtained a sufficient number of SNPs to cover almost all regions of their genomes by using RAD-seq analysis. The approach developed in this study would be applicable for mapping experiments that use crosses between closely related cultivars. However, for mapping with large populations (approx. 200 plants) it would be necessary to use other methods, such as genotype imputation (Andolfatto et al. 2011, Glaubitz et al. 2014), to estimate the genotype of each individual because the read coverage of a single locus in a single individual appears to be too low to estimate the genotype.
In this study, we identified two QTLs with RAD-seq BSA; the QTL on Gm10 coincided with the previously identified gene E2 (Watanabe et al. 2011), and the QTL on Gm 04 was close to the E8 locus (Cober et al. 2010). Because the sum of the genetic effects of E2 and the QTL on Gm04 is not sufficient to explain the broad-sense heritability of the flowering phenotype in TFF3 and the similarity between the flowering phenotypes of parental lines, other QTLs, which were not detected in our experiment, still segregated in TFF3 and we need to improve the RAD-seq BSA methods to enhance their power of QTL detection. One way to achieve this is to increase the population size before selecting plants for bulking. We choose 12 plants for bulking from a population consisting of 155 plants. The probability of having a homozygous allele in a single plant for one QTL is 3/8 in the F3 generation. The size of the current population was too small to obtain a plant harboring homozygous alleles for two or more QTLs. Increasing the population size would improve the detection power of BSA, but would also make the experiment more time- and labor-consuming. In addition, the resolution of BSA for the regions detected as QTLs depends on recombination events that have occurred in plants included in each bulk. The RAD-seq BSA developed in this study identified a major QTL in the 42.5–45.9-Mbp region on Gm10. The genetic length of this region was over 50 cM on the linkage map of Gm10 (Fig. 3B, Supplemental Table 2), indicating that the resolution of BSA alone is poor and further QTL analysis with the original TFF3 population and with additional DNA markers linked to the QTL is necessary to detect this QTL with a resolution of a few centimorgans. We consider the number of linked DNA markers obtained from RAD-seq BSA to be sufficient for introducing the target QTL into a different cultivar by marker-assisted selection.
The target region of the genome can be easily changed by changing the combination of restriction enzymes and the range of DNA fragment sizes (Supplemental Table 3). This flexibility would enhance the utility of RAD-seq BSA. Moreover, the number of loci detected by RAD-seq BSA seems to be comparable to that of other genotyping methods, such as SNP arrays in which SNPs positions are already known. Using the SNP array SoySNP50, Song et al. (2013) detected 47,337 polymorphic SNPs among 96 landraces, 96 elite cultivars from North America and 96 wild soybeans. The SoySNP50 array can analyze on average 9974 SNPs in a randomly selected pair of 96 elite cultivars. In this study, we used 7077 SNP loci to identify QTLs.
This study provides an example of identification of major genes controlling flowering time by RAD-seq BSA. We may need to improve our method. To analyze different traits in the same population, we need to prepare more than one DNA library, which can increase experimental costs. It would be difficult to apply our current method for identification of the loci underlying polygenic traits. Further study to extend the applicability of RAD-seq BSA to multiple traits (which can be analyzed by conventional QTL analysis) is necessary. Obtaining RAD-seq data from all individuals in a segregating population could improve the current strategy. To identify the gene responsible for the target QTL, QTL-seq proposed by Takagi et al. (2013) or a conventional positional cloning strategy needs to be applied. The methodology of this study, however, would be useful for the genetic mapping of mutants. A library developed by Tsuda et al. (2015) includes many soybean mutants not only for qualitative traits but also for quantitative traits such as protein and oil contents. The phenotypic changes in these mutants are expected to result from mutation(s) in a single or few genes. Identification of mutant gene(s) with strong effects on agronomic characteristics by RAD-seq BSA using populations derived from crosses between close relatives will provide novel breeding materials and DNA markers linked to the target loci.
Acknowledgements
We are grateful to Tokiko Kitajima for technical support. This study was supported by a grant from the Ministry of Agriculture, Forestry, and Fisheries of Japan (SFC1003).
Literature Cited
- Abe, A., S. Kosugi, K. Yoshida, S. Natsume, H. Takagi, H. Kanzaki, H. Matsumura, K. Yoshida, C. Mitsuoka, M. Tamiru et al. (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 30: 174–178.
- Andolfatto, P., D. Davison, D. Erezyilmaz, T.T. Hu, J. Mast, T. Sunayama-Morita and D.L. Stern (2011) Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Res. 21: 610–617.
- Baird, N.A., P.D. Etter, T.S. Atwood, M.C. Currey, A.L. Shiver, Z.A. Lewis, E.U. Selker, W.A. Cresko and E.A. Johnson (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3: e3376.
- Broman, K.W., H. Wu, S. Sen and G.A. Churchill (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19: 889–890.
- Camacho, C., G. Coulouris, V. Avagyan, N. Ma, J. Papadopoulos, K. Bealer and T.L. Madden (2009) BLAST+: architecture and applications. BMC Bioinformatics 10: 421.
- Cober, E.R., S.J. Molnar, M. Charette and H.D. Voldeng (2010) A new locus for early maturity in soybean. Crop Sci. 50: 524–527.
- Fehr, W.R., C.E. Caviness, D.T. Burmood and J.S. Pennington (1971) Stage of development descriptions for soybeans, Glycine Max (L.) Merrill. Crop Sci. 11: 929–931.
- Glaubitz, J.C., T.M. Casstevens, F. Lu, J. Harriman, R.J. Elshire, Q. Sun and E.S. Buckler (2014) TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 9: e90346.
- Hyten, D.L., Q. Song, Y. Zhu, I.Y. Choi, R.L. Nelson, J.M. Costa, J.E. Specht, R.C. Shoemaker and P.B. Cregan (2006) Impacts of genetic bottlenecks on soybean genome diversity. Proc. Natl. Acad. Sci. USA 103: 16666–16671.
- Hyten, D.L., I.Y. Choi, Q. Song, J.E. Specht, T.E. Carter, R.C. Shoemaker, E.Y. Hwang, L.K. Matukumalli and P.B. Cregan (2010) A high density integrated genetic linkage map of soybean and the development of a 1536 universal soy linkage panel for quantitative trait locus mapping. Crop Sci. 50: 960–968.
- Iwata, H. and S. Ninomiya (2006) AntMap: constructing genetic linkage maps using an ant colony optimization algorithm. Breed. Sci. 56: 371–377.
- Kaga, A., T. Shimizu, S. Watanabe, Y. Tsubokura, Y. Katayose, K. Harada, D.A. Vaughan and N. Tomooka (2012) Evaluation of soybean germplasm conserved in NIAS genebank and development of mini core collections. Breed. Sci. 61: 566–592.
- Katayose, Y., H. Kanamori, M. Shimomura, H. Ohyanagi, H. Ikawa, H. Minami, M. Shibata, T. Ito, K. Kurita, K. Ito et al. (2012) DaizuBase, an integrated soybean genome database including BAC-based physical maps. Breed. Sci. 61: 661–664.
- Kim, M.Y., S. Lee, K. Van, T.H. Kim, S.C. Jeong, I.Y. Choi, D.S. Kim, Y.S. Lee, D. Park, J. Ma et al. (2010) Whole-genome sequencing and intensive analysis of the undomesticated soybean (Glycine soja Sieb. and Zucc.) genome. Proc. Natl. Acad. Sci. USA 107: 22032–22037.
- Langmead, B. and S.L. Salzberg (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9: 357–359.
- Mitra, A., M. Skrzypczak, K. Ginalski and M. Rowicka (2015) Strategies for achieving high sequencing accuracy for low diversity samples and avoiding sample bleeding using Illumina platform. PLoS ONE 10: e0120520.
- Murray, M.G. and W.F. Thompson (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8: 4321–4325.
- Neff, M.M., J.D. Neff, J. Chory and A.E. Pepper (1998) dCAPS, a simple technique for the genetic analysis of single nucleotide polymorphisms: experimental applications in Arabidopsis thaliana genetics. Plant J. 14: 387–392.
- Neff, M.M., E. Turk and M. Kalishman (2002) Web-based primer design for single nucleotide polymorphism analysis. Trends Genet. 18: 613–615.
- Ohba, T. (1980) A new soybean cultivar “Fukuyutaka”. Agric. Technol. (Nogyo Gijutsu Japanese) 35: 511–513.
- Ohba, T. (1985) A new soybean cultivar “Toyoshirome”. Agric. Technol. (Nogyo Gijutsu Japanese) 40: 550–551.
- Poland, J.A., P.J. Brown, M.E. Sorrells, J.-L. Jannink, E. Paux, S. Faure, F. Choulet, D. Roger, V. Gauthier, E. Akhunov et al. (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7: e32253.
- Quarrie, S.A., V. Lazic-Jancic, D. Kovacevic, A. Steed and S. Pekic (1999) Bulk segregant analysis with molecular markers and its use for improving drought resistance in maize. J. Exp. Bot. 50: 1299–1306.
- R Development Core Team (2008) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org.
- Schmutz, J., S.B. Cannon, J. Schlueter, J. Ma, T. Mitros, W. Nelson, D.L. Hyten, Q. Song, J.J. Thelen, J. Cheng et al. (2010) Genome sequence of the palaeopolyploid soybean. Nature 463: 178–183.
- Shimomura, M., H. Kanamori, S. Komatsu, N. Namiki, Y. Mukai, K. Kurita, K. Kamatsuki, H. Ikawa, R. Yano, M. Ishimoto et al. (2015) The Glycine max cv. Enrei genome for improvement of Japanese soybean cultivars. Int. J. Genomics 2015: 358127.
- Song, Q.J., L.F. Marek, R.C. Shoemaker, K.G. Lark, V.C. Concibido, X. Delannay, J.E. Specht and P.B. Cregan (2004) A new integrated genetic linkage map of the soybean. Theor. Appl. Genet. 109: 122–128.
- Song, Q., D.L. Hyten, G. Jia, C.V. Quigley, E.W. Fickus, R.L. Nelson and P.B. Cregan (2013) Development and evaluation of SoySNP50K, a high-density genotyping array for soybean. PLoS ONE 8: e54985.
- Takagi, H., A. Abe, K. Yoshida, S. Kosugi, S. Natsume, C. Mitsuoka, A. Uemura, H. Utsushi, M. Tamiru, S. Takuno et al. (2013) QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 74: 174–183.
- Tsubokura, Y., S. Watanabe, Z. Xia, H. Kanamori, H. Yamagata, A. Kaga, Y. Katayose, J. Abe, M. Ishimoto and K. Harada (2014) Natural variation in the genes responsible for maturity loci E1, E2, E3 and E4 in soybean. Ann. Bot. 113: 429–441.
- Tsuda, M., A. Kaga, T. Anai, T. Shimizu, T. Sayama, K. Takagi, K. Machita, S. Watanabe, M. Nishimura, N. Yamada et al. (2015) Construction of a high-density mutant library in soybean and development of a mutant retrieval method using amplicon sequencing. BMC Genomics 16: 1014.
- Untergasser, A., I. Cutcutache, T. Koressaar, J. Ye, B.C. Faircloth, M. Remm and S.G. Rozen (2012) Primer3—new capabilities and interfaces. Nucleic Acids Res. 40: e115.
- Watanabe, S., Z. Xia, R. Hideshima, Y. Tsubokura, S. Sato, N. Yamanaka, R. Takahashi, T. Anai, S. Tabata, K. Kitamura et al. (2011) A map-based cloning strategy employing a residual heterozygous line reveals that the GIGANTEA gene is involved in soybean maturity and flowering. Genetics 188: 395–407.
- Zeng, Z.B. (1993) Theoretical basis for separation of multiple linked gene effects in mapping quantitative trait loci. Proc. Natl. Acad. Sci. USA 90: 10972–10976.
- Zhou, Z., Y. Jiang, Z. Wang, Z. Gou, J. Lyu, W. Li, Y. Yu, L. Shu, Y. Zhao, Y. Ma et al. (2015) Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 33: 408–414.
