Abstract
To explore the genetic resources that could be utilized to help improve root system architecture phenotypes in rice (Oryza sativa), we have conducted genome-wide association studies to investigate maximum root length and crown root number in 135 10-day-old Japanese rice accessions grown hydroponically. We identified a quantitative trait locus for crown root number at approximately 32.7 Mbp on chromosome 4 and designated it qNCR1 (quantitative trait locus for Number of Crown Root 1). A linkage disequilibrium map around qNCR1 suggested that three candidate genes are involved in crown root number: a cullin (LOC_Os04g55030), a gibberellin 20 oxidase 8 (LOC_Os04g55070), and a cyclic nucleotide-gated ion channel (LOC_Os04g55080). The combination of haplotypes for each gene was designated as a haploblock, and haploblocks 1, 2, and 3 were defined. Compared to haploblock 1, the accessions with haploblocks 2 and 3 had fewer crown roots; approximately 5% and 10% reductions in 10-day-old plants and 15% and 25% reductions in 42-day-old plants, respectively. A Japanese leading variety Koshihikari and its progenies harbored haploblock 3. Their crown root number could potentially be improved using haploblocks 1 and 2.
Introduction
Root system architecture (RSA) results from the deployment of various roots in the soil (Lynch 1995). In general, RSA can either help or limit the development of roots in the soil, which respectively, can positively or negatively affect plant growth (Gowariker et al. 2009, Lynch 1995). Rice (Oryza sativa) is one of the top three grain crops in the world, and several studies have shown that superior rice RSAs can result in increased yields in stressed soils. For example, rice productivity could be maintained under drought conditions if the depth of the root distribution was increased (Uga et al. 2013). While exposing the roots to the soil surface led to increased rice yields in saline paddies (Kitomi et al. 2020). Therefore, we assumed that there is an optimal rice RSA for each soil environment. The major challenge in RSA breeding is to develop new cultivars which can be adapted to different environments by improving RSAs using genetic resources (Uga 2021, Uga et al. 2015a).
Rice plants have fibrous root systems which consist of adventitious and lateral roots (Osmont et al. 2007), which are determined by three major components: root length, number, and angle. Root length, especially adventitious root length, affects the size of the RSA. An increase in maximum root length is related to the acquisition of water and nutrients (Courtois et al. 2009), and consequently, maximum root length is a target trait when breeding crops to efficiently use water and nutrient resources in the soil. Root number does not affect RSA size, but it does affect the root density. The higher the root density, the faster the absorption of water and nutrients (Marschner and Rengel 2012); which is further evidence by that fact in rice, root density and grain yield were found to be positively correlated (Yang et al. 2012). Therefore, root number is also a target when breeding to improve RSA. Root angle is the elongation angle of the roots, and is especially used to assess the adventitious roots in monocotyledons (Oyanagi 1994, Oyanagi et al. 1993, Trachsel et al. 2013, Uga et al. 2011). Changes in the root angle have a significant impact on the RSA as they affect the root distribution in the soil (Ge et al. 2000). Therefore, root angle modifications are widely targeted to improve the RSA in rice (Kitomi et al. 2020, Uga et al. 2013). Taken together, the combination of these three components should be targeted to improve RSA in rice.
Many quantitative trait loci (QTLs) responsible for RSA-related traits have been identified in rice. Previously, QTLs associated with root length (Courtois et al. 2009, Kitomi et al. 2018, Obara et al. 2014, Steele et al. 2006, Zhang et al. 2001), root number (Ali et al. 2000, Courtois et al. 2009, Islam et al. 2021, Ray et al. 1996), and root angle (Uga et al. 2011, 2013, 2015b) have all been identified. However, except for two genes associated with root angle (Kitomi et al. 2020, Uga et al. 2013), the genes responsible for most QTLs have not been isolated. Thus, the isolation of QTLs associated with root length and number is also required to further improve rice RSA.
Genome-wide association studies (GWAS) are powerful tools that can be utilized to identify the genes associated with agronomic traits; and the associations of nucleotide polymorphisms and phenotypic variances are best analyzed using a diverse population (Yano et al. 2016). Indeed, some RSA-related QTLs have previously been detected using GWAS in wheat and rice (Alemu et al. 2021, Beyer et al. 2019, Wang et al. 2019a), however, most were identified using bi-parental populations. GWAS generally narrow down the candidate genes inside linkage disequilibrium (LD) blocks. As the average size of LD blocks in cultivated rice is <500 kbp (Mather et al. 2007), we theorized that candidate genes in QTLs related to root length and number could be identified using GWAS.
In this study, using a GWAS mapping population that consisted of temperate japonica rice cultivars (Chigira et al. 2020), we performed GWAS to identify the QTL(s) associated with root length and number and evaluated the effects of the detected QTL(s) on phenotype. Furthermore, we investigated the geographic proportions in the different haploblocks and have discussed their potential applications.
Materials and Methods
Plant materials and cultivation
In this investigation 135 temperate japonica rice accessions, which were previously cultivated or used as parental breeding in Japan, were utilized (Chigira et al. 2020). As the population structure was low but there was a large phenotypic diversity, the accessions were suitable for a GWAS. Information on the accessions used, such as cultivar name and origin, was previously reported (Chigira et al. 2020).
To investigate maximum root length, crown root number, and shoot length at the early seedling stage of development, hydroponic cultivation with floating nets was performed. Seeds were surface sterilized with tap water containing fungicide at 15°C and immersed in tap water at 30°C for 2 d. The imbibed seeds were transferred onto a net floating on a 1/4-strength hydroponic solution based on Kimura B solution (Kitomi et al. 2018, Yoshida et al. 1976). The composition of the hydroponic solution was as follows: 91.25 μM (NH4)2SO4, 22.75 μM K2SO4, 136.75 μM MgSO4, 45.75 μM KNO3, 91.25 μM Ca(NO3)2, 45.5 μM KH2PO4, and 4.45 μM FeC6H5O7. The pH was adjusted to 5.5 with 5 mM MES (C6H13NO4S), HCl, and KOH. The hydroponic solution was changed every 3 or 4 d. Room temperature was defined as 30°C for 12 h during the day and 26°C for 12 h during the night in a greenhouse environment. Irradiation using 400 W metal-halide lamps was provided during the day. Poorly growing plants were thinned out prior to measurements. The seedlings were cultivated for 10 d in the greenhouse, and then their maximum root length, crown root number, and shoot length were measured. Data were collected from a maximum of ten individuals from each accession. Two cultivation trials were conducted.
To investigate crown root number in the late seedling stage of development, hydroponic cultivation with stainless-steel mesh baskets was performed. The mesh baskets (7.5 cm diameter × 5.0 cm depth) were filled with nutrient-poor soil Akadama (Ikubyo-Shibaue-Yodo, Shidara, Kanuma, Tochigi, Japan). Then, a germinated seed was sown at the center of each basket. Water and nutrients were supplied using a hydroponic solution: the 1/4- and 1/2-strength hydroponic solution, in which the pH was adjusted to 5.8 with HCl and KOH. The hydroponic solution was changed every 3 or 4 days. Room temperature and irradiation conditions were as defined previously for the maximum root length experiment. The seedlings were cultivated for six weeks in the greenhouse, after which their crown root numbers were counted. Data were collected from three individuals of each cultivar. Two cultivation trials were conducted.
Genome-wide association analysis
GWAS and gene-based association studies were performed according to a previous study (Chigira et al. 2020): we used a total of 670,069 SNPs and IndDels, and the haplotypes of 14,274 rice genes among the 135 temperate japonica rice cultivars. The linear mixed model incorporated the kinship data (Yu et al. 2006). A LD map was drawn using the “LDheatmap” R package (Shin et al. 2006) with the sequence variant data from the 135 accessions. The LD block was used to determine the candidate region.
Detection of the candidate genes
The criteria for the candidate genes was the same as the previous study (Chigira et al. 2020); the genes in the candidate region whose p value in the gene-based association study was <0.0001 in both the first and second trials were identified as candidate genes. A set of gene IDs and descriptions was downloaded from the Rice Annotation Project Database (Kawahara et al. 2013, Sakai et al. 2013).
Sequence logo analysis
Genomic sequences of a homologous gene in rice, Arabidopsis (Arabidopsis thalianla), Cyanidioschyzon merolae, and Saccharomyces cerevisiae were collected from the SALDA database version 3.0 (Mihara et al. 2009). We extracted dozens of amino acid sequences around the single nucleotide polymorphism position, and created a sequence logo using the “ggseqlogo” R package (Wagih 2017).
Analysis of geographical distribution of haplotypes
Origins of the accessions were grouped into nine region categories, which were all within Japan: Hokkaido, Tohoku, Kanto, Hokuriku, Tokai, Kinki, Shikoku, Chugoku, and Kyushu. The percentage of haplotypes in each category was drawn on a Japanese map using the “scatterpie” R package. The map was drawn using the “sf” R package (Pebesma 2018) with a shape file downloaded from “https://www.naturalearthdata.com/”.
Results
Phenotypic variations in the root traits of Japanese rice accessions
We cultivated 135 Japanese rice accessions hydroponically for 10 d after sowing to determine their maximum root length, root number, and shoot length (Fig. 1). For the maximum root length, a positive correlation between the first and second trials was observed (Fig. 1A) and there were no differences in the median values between the trials (Fig. 1B). This indicated that the differences in the first and second trials did not affect the maximum root length. For root number, a positive correlation between the first and second trials was observed (Fig. 1C), and the root number was slightly larger in the first trial (Fig. 1D). A similar tendency was observed in the results for shoot length (Fig. 1E, 1F), indicating that the first trial had a more positive effect on growth than the second. As positive correlation was observed between the three measured traits (Fig. 1A, 1C, 1E), we assumed that environmental differences were small in this study, and that the variations in the phenotypic data would reflect the genetic diversity of the population.

To identify the candidate genes affecting the root traits, a GWAS was conducted using the phenotypic data from the 135 rice cultivars. Manhattan plots for the maximum root length (Supplemental Fig. 1), root number (Fig. 2), and shoot length (Supplemental Fig. 2) are shown. We obtained one significant peak around 32.7 Mbp on chromosome 4 for root number (Fig. 2) and two significant peaks around 34.9 Mbp on chromosome 3 and 25.4 Mbp on chromosome 8 for shoot length (Supplemental Fig. 2) but none for maximum root length (Supplemental Fig. 1). The loci for the significant peaks associated with the shoot length and root number traits were different, which indicates that the candidate genes for root number and root length were not the same. The QTL for root number was named qNCR1 (quantitative trait locus for Number of Crown Root 1).
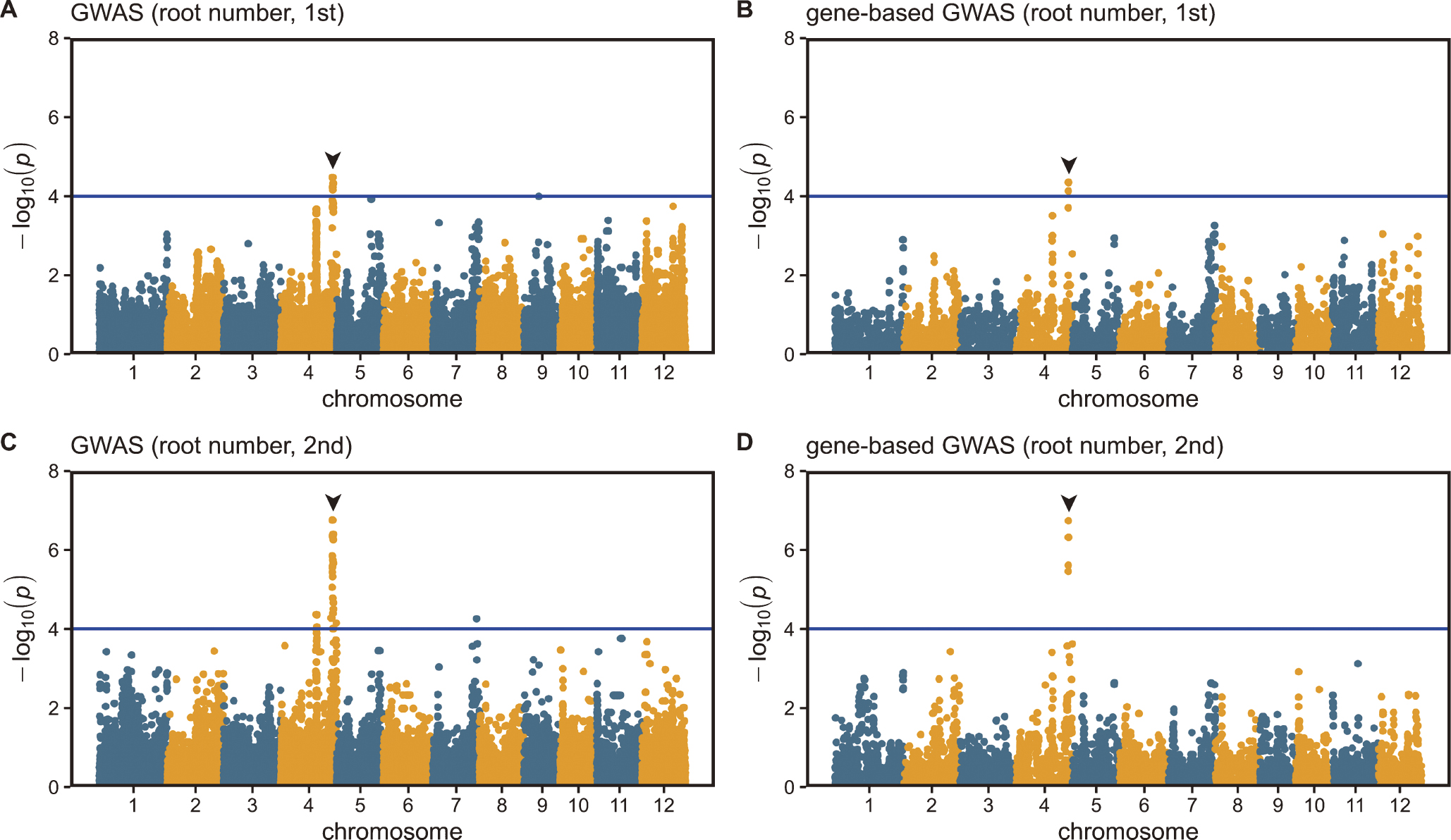
To identify the candidate genes affecting root number, we focused the Manhattan plot and LD map around qNCR1 (Fig. 3). Based on the LD heatmap (Fig. 3B), we delimited approximately 334-kbp candidate region (Fig. 3A). Genes with a p value <0.0001 in the gene-based GWAS are considered candidate genes, and this region had nine candidate genes [Table 1 (Ci et al. 2021, Gingerich et al. 2005, Nawaz et al. 2014)]. Excluding transposon-related genes, there were four candidate genes, namely LOC_Os04g55030, LOC_Os04g55070, LOC_Os04g55080, and LOC_Os04g55130. We investigated the expression profile of them by RiceXPro database (Sato et al. 2011a, 2011b, 2013) and found that LOC_Os04g55030 and LOC_Os04g55080 were expressed ubiquitously in all tissues including roots and LOC_Os04g55070 and LOC_Os04g55130 were highly expressed in roots and inflorescence, respectively (Supplemental Figs. 3–6). Focusing on genes that are highly expressed in the roots, three annotated genes were left as candidates: a cullin (LOC_Os04g55030), a gibberellin (GA) 20 oxidase (GA20ox, LOC_Os04g55070), and a cyclic nucleotide-gated ion channel (CNGC, LOC_Os04g55080). All three candidates had missense mutations in the GWAS population (Supplemental Fig. 7). Among them, LOC_Os04g55030 and LOC_Os04g55070 showed a change in the charge of the amino acid (Supplemental Fig. 7A, 7B). Sequence logo analysis revealed that the 223rd amino acid of LOC_Os04g55030 was highly conserved as an amino acid with a negative charge among the 51 homologous sequences (Supplemental Fig. 8), suggesting that a change from a negative to a positive charge, would influence its function. However, as any gene in the LD block could be a candidate gene, we could not indicate the causal gene for root number.

Table 1.
Candidate genes associated with root number
| MSU ID |
Gene |
–Log10(p) |
Description |
Reference |
| LOC_Os04g55140 |
|
6.44 |
Retrotransposon protein, putative, Ty1-copia subclass, expressed |
|
| LOC_Os04g55030 |
OsCUL3b |
6.40 |
Cullin, putative, expressed |
(Gingerich et al. 2005) |
| LOC_Os04g55080 |
OsCNGC8 |
6.40 |
Cyclic nucleotide-gated ion channel, putative, expressed |
(Nawaz et al. 2014) |
| LOC_Os04g55130 |
|
6.40 |
Expressed protein |
|
| LOC_Os04g55350 |
|
6.40 |
Retrotransposon protein, putative, Ty3-gypsy subclass, expressed |
|
| LOC_Os04g55450 |
|
6.40 |
Transposon protein, putative, unclassified, expressed |
|
| LOC_Os04g55540 |
|
6.40 |
Retrotransposon protein, putative, unclassified, expressed |
|
| LOC_Os04g55020 |
|
5.40 |
Retrotransposon protein, putative, Ty3-gypsy subclass, expressed |
|
| LOC_Os04g55070 |
OsGA20ox8 |
4.16 |
Gibberellin 20 oxidase 2, putative, expressed |
(Ci et al. 2021) |
Based on the variant call results in the coding sequences, we detected two haplotypes in LOC_Os04g55030, LOC_Os04g55070, and LOC_Os04g55080 (Supplemental Fig. 7); haplotype 1, which is identical to the RAP-DB sequence (Kawahara et al. 2013, Sakai et al. 2013), and haplotype 2. Haplotypes of LOC_Os04g55030 and LOC_Os04g55080 were completely linked. To investigate the influence of haplotypes 1 and 2 on root number, we compared the root numbers from the 135 rice cultivars 10 d (Supplemental Fig. 9A, 9B) and 42 d after sowing (Supplemental Fig. 9C, 9D). The group harboring haplotype 2 had significantly lower root numbers when compared to the group harboring haplotype 1 in all conditions (Supplemental Fig. 9). The reduction in the number of roots was approximately 10% 10 d and 25% 42 d after sowing (Supplemental Fig. 9).
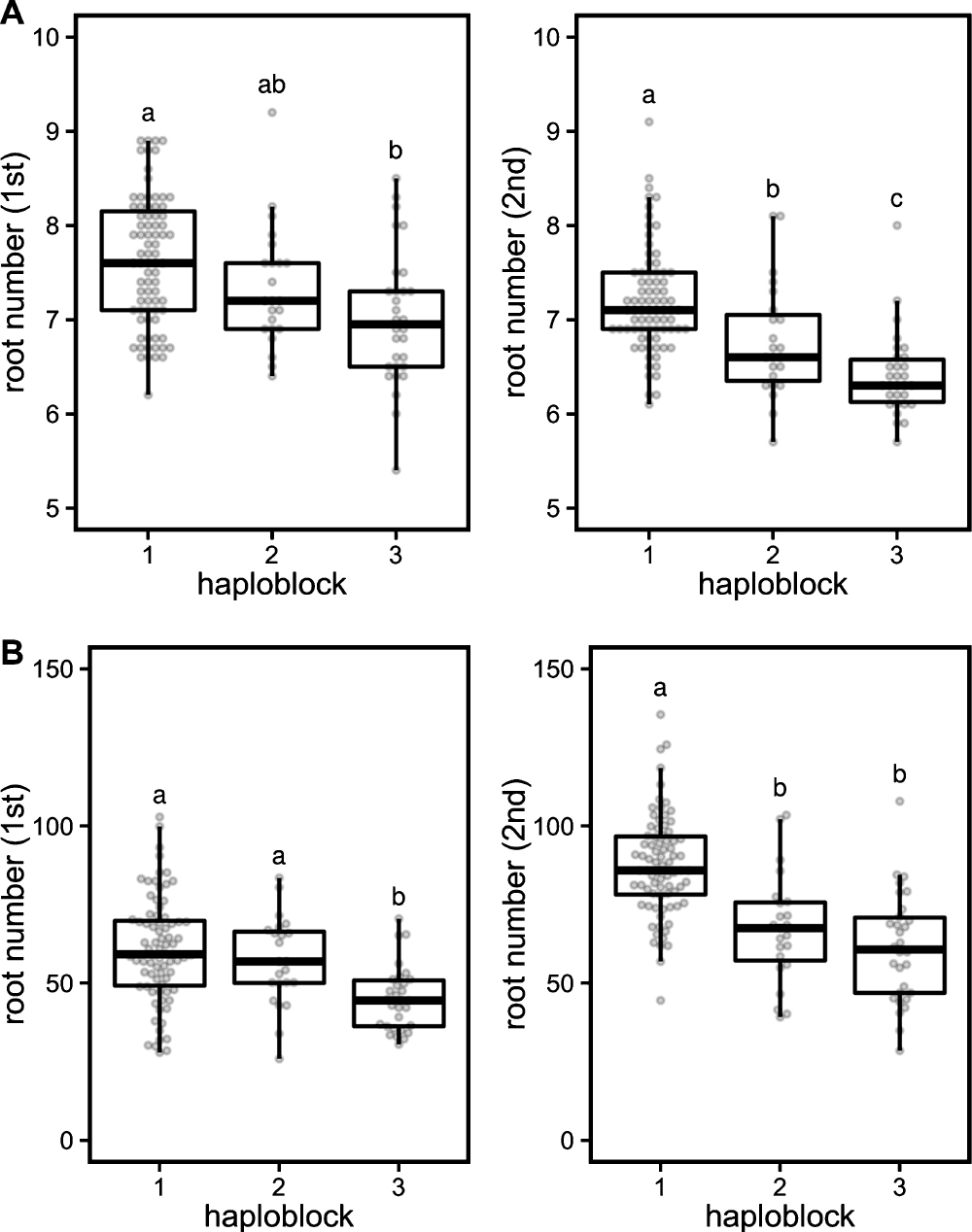
The combination of the haplotypes for each gene were designated as a haploblock, and haploblocks 1, 2, and 3 were defined. Haploblock 1 harbored haplotype 1 at all genes. Haploblock 2 harbored haplotype 1 at LOC_Os04g55030 and LOC_Os04g55080 and haplotype 2 at LOC_Os04g55070. Haploblock 3 harbored haplotype 2 at all genes. When compared to haploblock 1, the accessions with haploblocks 2 and 3 had fewer crown roots, with approximately 5% and 10% reductions in the 10 d old plants and 15% and 25% reductions in the 42 d old plants, respectively (Fig. 4). Haploblock 3 reproducibly reduced the crown root number, while haploblock 2 did not (Fig. 4).
Distribution of the three haploblocks
To investigate the distribution of the three haploblocks, we obtained genomic sequences of 69 accessions from the world rice core collection (WRC) and 50 accessions from the rice core collection of Japanese landraces [JRC; https://ricegenome-corecollection.dna.affrc.go.jp/) (Tanaka et al. 2020, 2021)]. Haploblock 2 was distributed among almost all world-wide accessions whereas haploblocks 1 and 3 were mainly distributed in the Japanese accessions (Supplemental Table 1). A Japanese leading variety Koshihikari (Kobayashi et al. 2018) and its progenies harbored haploblock 3, implying that haploblock 3 was spread throughout Japan during breeding processes that utilized Koshihikari. We investigated a more detailed study of the distribution of the three haploblocks in Japan using the rice accession sequences that had been used in the GWAS (Fig. 5). We found that haploblocks 2 and 3 were concentrated in the Hokkaido and Hokuriku regions in Japan, respectively (Fig. 5). Hokkaido is the northernmost region of Japan and is one of the northern limits for rice cultivation (Fujino et al. 2019). Hokuriku is the region where the Koshihikari cultivar was first developed (Kobayashi et al. 2018).

Discussion
This study detected a QTL for crown root number, qNCR1, for which three possible candidate genes were also identified, a cullin (LOC_Os04g55030), a GA20ox (LOC_Os04g55070), and a CNGC (LOC_Os04g55080), although any gene in the LD block could be a candidate. The cullin protein assembles multi-subunit Cullin-RING E3 ubiquitin ligase complexes, involved in various functions, such as cell-cycle control, DNA replication, and development (Sarikas et al. 2011). GA20ox is a GA biosynthesis enzyme, involved in many aspects of plant growth and development (Spielmeyer et al. 2002). CNGC is a nonselective cation channel (Kaupp and Seifert 2002). We defined three haploblocks based on their combinations of the haplotypes for LOC_Os04g55030, LOC_Os04g55070, and LOC_Os04g55080. Accessions harboring haploblocks 1 and 3 had the highest and the lowest crown root numbers, respectively (Fig. 4). The root numbers in accessions with haploblocks 2 and 3, when compared to those with haploblock 1, were approximately 5% and 10% lower in the 10 d old plants and 15% and 25% lower in the 42 d old plants, respectively. This indicated that the root number could be improved exchanging these haploblocks in qNCR1. Because haploblock 1 harbored haplotype 1 at all genes, and haploblock 2 harbored haplotype 1 at LOC_Os04g55030 and LOC_Os04g55080 and haplotype 2 at LOC_Os04g55070, at least LOC_Os04g55070 might affect the crown root number. Furthermore, haploblock 3 harbored haplotype 2 at all genes, indicating that LOC_Os04g55030 and/or LOC_Os04g55080 might affect the crown root number additively with LOC_Os04g55070. Therefore, we assumed that at least two genes associated with the crown root number.
LOC_Os04g55030, is a CULLIN3 (CUL3) gene family member, OsCUL3b (Gingerich et al. 2005). CUL3 is a component of CUL3 E3 ligases, and it binds target substrates via “Bric a brac, Tramtrack and Broad Complex/Pox virus, and Zinc finger” (BTB/POZ) proteins and it binds a ubiquitin conjugating enzyme, E2 ligase, resulting in the ubiquitination of a target sequence (Ban and Estelle 2021). As 80 and 149 BTB-domain proteins are present in the Arabidopsis and rice genomes, respectively, and the majority of them interact with CUL3 (Ban and Estelle 2021), these complexes may patriciate in various regulations of plant development. Arabidopsis genomes contain two CUL3 genes, AtCUL3a and AtCUL3b. It was reported that mutations in both AtCUL3a and AtCUL3b resulted in embryo development arrest (Figueroa et al. 2005) and that AtCUL3a is involved in regulating immunity by degrading AtNPR1, a homolog of OsNPR1 (Spoel et al. 2009). These results indicated that CUL3s are involved in essential development processes and immune responses. Furthermore, AtCUL3a interacts with ETO1, a BTB-domain protein which directly interacts with the rate-limiting enzyme in ethylene biosynthesis 1-aminocyclopropane-1-carboxylate synthase 5 (ACS5), to degrade ACS5 (Wang et al. 2004). A CUL3 hypomorphic mutant failed to degrade ACS5 and exhibits a constitutive ethylene response (Thomann et al. 2009). The rice genome contains three CUL3 genes, OsCUL3a, OsCUL3b, and OsCUL3c (Gingerich et al. 2005), of which the analysis of OsCUL3a, which is involved in cell death and immune responses, is the most advanced (Gao et al. 2020, Liu et al. 2017). It is reported that mutations in OsCUL3a result in a severe cell death phenotype at the tillering stage and enhanced resistance to pathogens by degrading OsNPR1, a positive regulator of cell death in rice, via the 26S proteasome (Liu et al. 2017). There is no previous evidence that OsCUL3b influences crown root number, but we considered that OsCUL3b could be involved with crown root number as adequate auxin signaling is important for crown root emergence and CUL3 potentially interacts with BTB-domain proteins, some of which reportedly participate in auxin-mediated plant development (Cheng et al. 2007, Mandadi et al. 2009).
LOC_Os04g55070 is OsGA20ox8, an enzyme that is involved in bioactive GAs synthesis (Ci et al. 2021). The rice genome contains eight OsGA20ox genes, OsGA20ox1–8 (Ci et al. 2021, Han and Zhu 2011). The most well-known GA20ox gene is GA20ox2, as its loss-of-function allele was used in the green revolution (Ashikari et al. 2002). GA20ox4 is involved in panicle length, as higher expression levels of GA20ox4 in young panicles results in panicle rachis elongation (Agata et al. 2020). OsGA20ox1 (Oikawa et al. 2004) and OsGA20ox3 (Qin et al. 2013) are also involved in GAs synthesis. OsGA20ox6 was reported to be a dioxygenase that converts indole-3-acetic acid (IAA) into inactive OxIAA (Zhao et al. 2013), indicating that not all OsGA20oxs are involved in GA metabolism (Liu et al. 2020). Another example is OsGA20ox7, which is involved in salicylic acid homeostasis (Liu et al. 2020). As mentioned above, GAs are involved in many aspects of plant growth and development (Spielmeyer et al. 2002), and changes in their levels also influence adventitious root formation (Lo et al. 2008, Mauriat et al. 2014). Although the roles of GA20ox8 (LOC_Os04g55070) are currently unclear, GA20ox8 may influence crown root number.
LOC_Os04g55080 is OsCNGC8 (Nawaz et al. 2014), which is a member of CNGC encoding a nonselective cation channel permeable to cations such as K+, Na+, and Ca2+ (Kaupp and Seifert 2002). The rice genome contains sixteen OsCNGC genes, OsCNGC1–16 (Nawaz et al. 2014). OsCNGC1 contributed to salt stress tolerance as they can help to avert toxic Na+ influxes (Assaha et al. 2017). OsCNGC13 is permeable to Ca2+ to facilitate pollen tube growth (Xu et al. 2017). OsCNGC9 plays an important role in the resistance to rice blast disease by mediating pathogen-associated molecular pattern (PAMP)-induced Ca2+ influxes (Wang et al. 2019b). OsCNGC14 and OsCNGC16 are involved in the tolerance to heat and chilling stresses and their loss-of -function can reduce or abolish cytosolic calcium signals that are induced by either heat or chilling stresses (Cui et al. 2020). As shown above, the CNGC family has a wide range of functions, from simple ion transport to the regulation of calcium signaling. It is possible that there are functions in the CNGC family that affect root number, but the specific functions of OsCNGC8 are still unknown.
Taken together, all three candidate genes, LOC_Os04g55030, LOC_Os04g55070, and LOC_Os04g55080, are potentially causal gene(s) for qNCR1. To confirm the identity of the genes involved, it will be necessary to make knockout mutants by CRISPR/Cas9 system and/or conduct complementation tests in the future. It seems, however, that it may be possible to improve root systems by using haploblocks of qNCR1 even though the causal genes were not confirmed in this study. When compared to haploblock 1, haploblocks 2 and 3 appeared to effectively reduce the number of roots (Fig. 4). The distribution map of each haploblock in Japan showed some bias; as haploblock 1 was rarely identified in the Hokkaido region while haploblocks 2 and 3 appeared with a high frequency in the Hokkaido and Hokuriku regions, respectively (Fig. 5). Hokkaido is the northernmost region of Japan and is one of the northern limits for rice cultivation (Fujino et al. 2019). We assumed that haploblock 2 in the Hokkaido variety was derived from a landrace Akage (Supplemental Table 2), which is a cultivar that was used in early breeding programs at Hokkaido (Fujino et al. 2019). Several QTLs involved in heading date (Fujino et al. 2019, Yokoo et al. 1980) and cold tolerance (Saito et al. 2001, 2004) were used to help adapt rice to the Hokkaido environment. A cold tolerance QTL Ctb1 was previously located at approximately 31.8 Mbp on chromosome 4 (Saito et al. 2004, Yonemaru et al. 2010), which was close to the location of qNCR1 (32.7 Mbp on chromosome 4, Fig. 2). Therefore, we assumed that qNCR1 was spread via the breeding program that focused on cold tolerance via linkage drag. The Hokuriku region is also where the Koshihikari cultivar was developed (Kobayashi et al. 2018). Since haploblock 3 was distributed only in a small number of cultivars including Koshihikari and Koshihikari-derived cultivars in Japan, we assumed that the cultivars with haploblock 3 have not been used after recent breeding using Koshihikari. We found that Morita-wase (landrace), an ancestor of Koshihikari (Kobayashi et al. 2018), harbored haploblock 3 (Supplemental Table 2). Therefore, we assumed that haploblock 3 in Morita-wase was inherited by Koshihikari and Koshihikari-derived cultivars.
The tiller number has a strong influence on root number (Kawakatsu et al. 2021). Because qNCR1 was detected in juvenile seedlings with no tillering (Fig. 2A, 2B), we assumed that qNCR1 did not influence the tiller number at juvenile stage. However, influence of qNCR1 on tiller number at adult stage were not elucidated. Although we need further investigation to elucidate it before qNCR1 will be used as breeding materials, these haploblocks of qNCR1 could be potentially used to improve crown root number traits in rice. Koshihikari and Koshihikari-derived cultivars harbored haploblock 3, indicating that these cultivars could use haploblocks 1 and 2 to increase the root number. Almost all world-wide cultivars harbored haploblock 2 (Supplemental Table 1), indicating that these cultivars could use haploblocks 1 and 3 to increase and reduce the root number, respectively. The root number is associated with root density which is compatible with water and nutrient uptake (Marschner and Rengel 2012). Therefore, increasing the number of roots may improve the nutrient absorption characteristics of the crop. However, in some environments such as low water conditions, crops with reduced root numbers can perform better (Gao and Lynch 2016). Consequently, we propose that these haploblocks of qNCR1 could be used to breed rice cultivars that are adapted to specific environments.
Author Contribution Statement
ST designed the study, obtained the trait data at young growth period, analyzed all data, and wrote the manuscript. MY provided the SNP and indel data, supervised and conducted the GWAS. YU coordinated the project, designed the study, obtained the trait data at a middle growth period, and revised the manuscript.
Acknowledgments
We thank Yuka Kitomi, Satoko Takayasu, Natsumi Maruyama, Yoko Fukuda, and Emiko Odajima for their experimental assistant. This study was supported by a grant from the Ministry of Agriculture, Forestry and Fisheries of Japan [Smart breeding system for Innovative Agriculture (BAC2001)] and the Japan Science and Technology Agency (JST) CREST Grant Number JPMJCR17O1 and JPMJCR17O3.
Literature Cited
- Agata, A., K. Ando, S. Ota, M. Kojima, Y. Takebayashi, S. Takehara, K. Doi, M. Ueguchi-Tanaka, T. Suzuki, H. Sakakibara et al. (2020) Diverse panicle architecture results from various combinations of Prl5/GA20ox4 and Pbl6/APO1 alleles. Commun Biol 3: 302.
- Alemu, A., T. Feyissa, M. Maccaferri, G. Sciara, R. Tuberosa, K. Ammar, A. Badebo, M. Acevedo, T. Letta and B. Abeyo (2021) Genome-wide association analysis unveils novel QTLs for seminal root system architecture traits in Ethiopian durum wheat. BMC Genomics 22: 20.
- Ali, M.L., M.S. Pathan, J. Zhang, G. Bai, S. Sarkarung and H.T. Nguyen (2000) Mapping QTLs for root traits in a recombinant inbred population from two indica ecotypes in rice. Theor Appl Genet 101: 756–766.
- Ashikari, M., A. Sasaki, M. Ueguchi-Tanaka, H. Itoh, A. Nishimura, S. Datta, K. Ishiyama, T. Saito, M. Kobayashi, G.S. Khush et al. (2002) Loss-of-function of a rice gibberellin biosynthetic gene, GA20 oxidase (GA20ox-2), led to the rice “green revolution”. Breed Sci 52: 143–150.
- Assaha, D.V.M., A. Ueda, H. Saneoka, R. Al-Yahyai and M.W. Yaish (2017) The role of Na+ and K+ transporters in salt stress adaptation in glycophytes. Front Physiol 8: 509.
- Ban, Z. and M. Estelle (2021) CUL3 E3 ligases in plant development and environmental response. Nat Plants 7: 6–16.
- Beyer, S., S. Daba, P. Tyagi, H. Bockelman, G. Brown-Guedira, IWGSC and M. Mohammadi (2019) Loci and candidate genes controlling root traits in wheat seedlings—a wheat root GWAS. Funct Integr Genomics 19: 91–107.
- Cheng, Y., G. Qin, X. Dai and Y. Zhao (2007) NPY1, a BTB-NPH3-like protein, plays a critical role in auxin-regulated organogenesis in Arabidopsis. Proc Natl Acad Sci USA 104: 18825–18829.
- Chigira, K., N. Kojima, M. Yamasaki, K. Yano, S. Adachi, T. Nomura, M. Jiang, K. Katsura and T. Ookawa (2020) Landraces of temperate japonica rice have superior alleles for improving culm strength associated with lodging resistance. Sci Rep 10: 19855.
- Ci, J., X. Wang, Q. Wang, F. Zhao, W. Yang, X. Cui, L. Jiang, X. Ren and W. Yang (2021) Genome-wide analysis of gibberellin-dioxygenases gene family and their responses to GA applications in maize. PLoS One 16: 5.
- Courtois, B., N. Ahmadi, F. Khowaja, A.H. Price, J.F. Rami, J. Frouin, C. Hamelin and M. Ruiz (2009) Rice root genetic architecture: Meta-analysis from a drought QTL database. Rice (N Y) 2: 115–128.
- Cui, Y., S. Lu, Z. Li, J. Cheng, P. Hu, T. Zhu, X. Wang, M. Jin, X. Wang, L. Li et al. (2020) CYCLIC NUCLEOTIDE-GATED ION CHANNELs 14 and 16 promote tolerance to heat and chilling in rice. Plant Physiol 183: 1794–1808.
- Figueroa, P., G. Gusmaroli, G. Serino, J. Habashi, L. Ma, Y. Shen, S. Feng, M. Bostick, J. Callis, H. Hellmann et al. (2005) Arabidopsis has two redundant cullin3 proteins that are essential for embryo development and that interact with RBX1 and BTB proteins to form multisubunit E3 ubiquitin ligase complexes in vivo. Plant Cell 17: 1180–1195.
- Fujino, K., Y. Hirayama and R. Kaji (2019) Marker-assisted selection in rice breeding programs in Hokkaido. Breed Sci 69: 383–392.
- Gao, Y. and J.P. Lynch (2016) Reduced crown root number improves water acquisition under water deficit stress in maize (Zea mays L.). J Exp Bot 67: 4545–4557.
- Gao, Z., Q. Liu, Y. Zhang, D. Chen, X. Zhan, C. Deng, S. Cheng and L. Cao (2020) Oscul3a-associated molecular switches have functions in cell metabolism, cell death, and disease resistance. J Agric Food Chem 68: 5471–5482.
- Ge, Z., G. Rubio and J.P. Lynch (2000) The importance of root gravitropism for inter-root competition and phosphorus acquisition efficiency: Results from a geometric simulation model. Plant Soil 218: 159–171.
- Gingerich, D.J., J.M. Gagne, D.W. Salter, H. Hellmann, M. Estelle, L. Ma and R.D. Vierstra (2005) Cullins 3a and 3b assemble with members of the broad complex/tramtrack/bric-a-brac (BTB) protein family to form essential ubiquitin-protein ligases (E3s) in Arabidopsis. J Biol Chem 280: 18810–18821.
- Gowariker, V., V.N. Krishnamurthy, S. Gowariker, M. Dhanorkar and K. Paranjape (2009) The fertilizer encyclopedia. John Wiley & Sons., Hoboken, New Jersey.
- Han, F. and B. Zhu (2011) Evolutionary analysis of three gibberellin oxidase genesin rice, Arabidopsis, and soybean. Gene 473: 23–35.
- Islam, A., Y. Zhang, G. Anis, M.H. Rani, W. Anley, Q. Yang, L. Liu, X. Shen, L. Cao, S. Cheng et al. (2021) Fine mapping and candidate gene analysis of qRN5a, a novel QTL promoting root number in rice under low potassium. Theor Appl Genet 134: 213–227.
- Kaupp, U.B. and R. Seifert (2002) Cyclic nucleotide-gated ion channels. Physiol Rev 82: 769–824.
- Kawahara, Y., M. de la Bastide, J.P. Hamilton, H. Kanamori, W.R. Mccombie, S. Ouyang, D.C. Schwartz, T. Tanaka, J. Wu, S. Zhou et al. (2013) Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice (N Y) 6: 4.
- Kawakatsu, T., S. Teramoto, S. Takayasu, N. Maruyama, R. Nishijima, Y. Kitomi and Y. Uga (2021) The transcriptomic landscapes of rice cultivars with diverse root system architectures grown in upland field conditions. Plant J 106: 1177–1190.
- Kitomi, Y., E. Nakao, S. Kawai, N. Kanno, T. Ando, S. Fukuoka, K. Irie and Y. Uga (2018) Fine mapping of QUICK ROOTING 1 and 2, quantitative trait loci increasing root length in rice. G3 (Bethesda) 8: 727–735.
- Kitomi, Y., E. Hanzawa, N. Kuya, H. Inoue, N. Hara, S. Kawai, N. Kanno, M. Endo, K. Sugimoto, T. Yamazaki et al. (2020) Root angle modifications by the DRO1 homolog improve rice yields in saline paddy fields. Proc Natl Acad Sci USA 117: 21242–21250.
- Kobayashi, A., K. Hori, T. Yamamoto and M. Yano (2018) Koshihikari: A premium short-grain rice cultivar—its expansion and breeding in Japan. Rice (N Y) 11: 15.
- Liu, Q., Y. Ning, Y. Zhang, N. Yu, C. Zhao, X. Zhan, W. Wu, D. Chen, X. Wei, G.L. Wang et al. (2017) OsCUL3a negatively regulates cell death and immunity by degrading OsNPR1 in rice. Plant Cell 29: 345–359.
- Liu, X., W.J. Cai, X. Yin, D. Yang, T. Dong, Y.Q. Feng and Y. Wu (2020) Two SLENDER and CRINKLY LEAF dioxygenases play an essential role in rice shoot development. J Exp Bot 71: 1387–1401.
- Lo, S.F., S.Y. Yang, K.T. Chen, Y.I. Hsing, J.A.D. Zeevaart, L.J. Chen and S.M. Yu (2008) A novel class of gibberellin 2-oxidases control semidwarfism, tillering, and root development in rice. Plant Cell 20: 2603–2618.
- Lynch, J. (1995) Root architecture and plant productivity. Plant Physiol 109: 7–13.
- Mandadi, K.K., A. Misra, S. Ren and T.D. Mcknight (2009) BT2, a BTB protein, mediates multiple responses to nutrients, stresses, and hormones in Arabidopsis. Plant Physiol 150: 1930–1939.
- Marschner, P. and Z. Rengel (2012) Nutrient availability in soils. In: Marschner, P. (ed.) Marschner’s mineral nutrition of higher plants. third. Elsevier, Amsterdam, pp. 315–330.
- Mather, K.A., A.L. Caicedo, N.R. Polato, K.M. Olsen, S. McCouch and M.D. Purugganan (2007) The extent of linkage disequilibrium in rice (Oryza sativa L.). Genetics 177: 2223–2232.
- Mauriat, M., A. Petterle, C. Bellini and T. Moritz (2014) Gibberellins inhibit adventitious rooting in hybrid aspen and Arabidopsis by affecting auxin transport. Plant J 78: 372–384.
- Mihara, M., T. Itoh and T. Izawa (2009) SALAD database: A motif-based database of protein annotations for plant comparative genomics. Nucleic Acids Res 38: 835–842.
- Nawaz, Z., K.U. Kakar, M.A. Saand and Q.Y. Shu (2014) Cyclic nucleotide-gated ion channel gene family in rice, identification, characterization and experimental analysis of expression response to plant hormones, biotic and abiotic stresses. BMC Genomics 15: 853.
- Obara, M., T. Ishimaru, T. Abiko, D. Fujita, N. Kobayashi, S. Yanagihara and Y. Fukuta (2014) Identification and characterization of quantitative trait loci for root elongation by using introgression lines with genetic background of indica-type rice variety IR64. Plant Biotechnol Rep 8: 267–277.
- Oikawa, T., M. Koshioka, K. Kojima, H. Yoshida and M. Kawata (2004) A role of OsGA20ox1, encoding an isoform of gibberellin 20-oxidase, for regulation of plant stature in rice. Plant Mol Biol 55: 687–700.
- Osmont, K.S., R. Sibout and C.S. Hardtke (2007) Hidden branches: Developments in root system architecture. Annu Rev Plant Biol 58: 93–113.
- Oyanagi, A. (1994) Gravitropic response growth angle and vertical distribution of roots of wheat (Triticum aestivum L.). Plant Soil 165: 323–326.
- Oyanagi, A., T. Nakamoto and M. Wada (1993) Relationship between root growth angle of seedlings and vertical distribution of roots in the field in wheat cultivars. Jpn J Crop Sci 62: 565–570.
- Pebesma, E. (2018) Simple features for R: Standardized support for spatial vector data. R J 10: 439–446.
- Qin, X., J.H. Liu, W.S. Zhao, X.J. Chen, Z.J. Guo and Y.L. Peng (2013) Gibberellin 20-oxidase gene OsGA20ox3 regulates plant stature and disease development in rice. Mol Plant Microbe Interact 26: 227–239.
- Ray, J.D., L. Yu, S.R. McCouch, M.C. Champoux, G. Wang and H.T. Nguyen (1996) Mapping quantitative trait loci associated with root penetration ability in rice (Oryza sativa L.). Theor Appl Genet 92: 627–636.
- Saito, K., K. Miura, K. Nagano, Y. Hayano-Saito, H. Araki and A. Kato (2001) Identification of two closely linked quantitative trait loci for cold tolerance on chromosome 4 of rice and their association with anther length. Theor Appl Genet 103: 862–868.
- Saito, K., Y. Hayano-Saito, W. Maruyama-Funatsuki, Y. Sato and A. Kato (2004) Physical mapping and putative candidate gene identification of a quantitative trait locus Ctb1 for cold tolerance at the booting stage of rice. Theor Appl Genet 109: 515–522.
- Sakai, H., S.S. Lee, T. Tanaka, H. Numa, J. Kim, Y. Kawahara, H. Wakimoto, C.C. Yang, M. Iwamoto, T. Abe et al. (2013) Rice annotation project database (RAP-DB): An integrative and interactive database for rice genomics. Plant Cell Physiol 54: e6.
- Sarikas, A., T. Hartmann and Z.Q. Pan (2011) The cullin protein family. Genome Biol 12: 1–12.
- Sato, Y., B. Antonio, N. Namiki, R. Motoyama, K. Sugimoto, H. Takehisa, H. Minami, K. Kamatsuki, M. Kusaba, H. Hirochika et al. (2011a) Field transcriptome revealed critical developmental and physiological transitions involved in the expression of growth potential in japonica rice. BMC Plant Biol 11: 10.
- Sato, Y., B.A. Antonio, N. Namiki, H. Takehisa, H. Minami, K. Kamatsuki, K. Sugimoto, Y. Shimizu, H. Hirochika and Y. Nagamura (2011b) RiceXPro: A platform for monitoring gene expression in japonica rice grown under natural field conditions. Nucleic Acids Res 39: 1141–1148.
- Sato, Y., H. Takehisa, K. Kamatsuki, H. Minami, N. Namiki, H. Ikawa, H. Ohyanagi, K. Sugimoto, B.A. Antonio and Y. Nagamura (2013) RiceXPro Version 3.0: Expanding the informatics resource for rice transcriptome. Nucleic Acids Res 41: 1206–1213.
- Shin, J.-H., S. Blay, B. McNeney and J. Graham (2006) LDheatmap: An R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J Stat Softw 16: 3.
- Spielmeyer, W., M.H. Ellis and P.M. Chandler (2002) Semidwarf (sd-1), “green revolution” rice, contains a defective gibberellin 20-oxidase gene. Proc Natl Acad Sci USA 99: 9043–9048.
- Spoel, S.H., Z. Mou, Y. Tada, N.W. Spivey, P. Genschik and X. Dong (2009) Proteasome-mediated turnover of the transcription coactivator NPR1 plays dual roles in regulating plant immunity. Cell 137: 860–872.
- Steele, K.A., A.H. Price, H.E. Shashidhar and J.R. Witcombe (2006) Marker-assisted selection to introgress rice QTLs controlling root traits into an Indian upland rice variety. Theor Appl Genet 112: 208–221.
- Tanaka, N., M. Shenton, Y. Kawahara, M. Kumagai, H. Sakai, H. Kanamori, J. Yonemaru, S. Fukuoka, K. Sugimoto, M. Ishimoto et al. (2020) Whole-Genome Sequencing of the NARO World Rice Core Collection (WRC) as the basis for diversity and association studies. Plant Cell Physiol 61: 922–932.
- Tanaka, N., M. Shenton, Y. Kawahara, M. Kumagai, H. Sakai, H. Kanamori, J.I. Yonemaru, S. Fukuoka, K. Sugimoto, M. Ishimoto et al. (2021) Investigation of the genetic diversity of a Rice Core Collection of Japanese landraces using whole-genome sequencing. Plant Cell Physiol 61: 2087–2096.
- Thomann, A., E. Lechner, M. Hansen, E. Dumbliauskas, Y. Parmentier, J. Kieber, B. Scheres and P. Genschik (2009) Arabidopsis CULLIN3 genes regulate primary root growth and patterning by ethylene-dependent and -independent mechanisms. PLoS Genet 5: e1000328.
- Trachsel, S., S.M. Kaeppler, K.M. Brown and J.P. Lynch (2013) Maize root growth angles become steeper under low N conditions. Field Crops Res 140: 18–31.
- Uga, Y. (2021) Challenges to design-oriented breeding of root system architecture adapted to climate change. Breed Sci 71: 3–12.
- Uga, Y., K. Okuno and M. Yano (2011) Dro1, a major QTL involved in deep rooting of rice under upland field conditions. J Exp Bot 62: 2485–2494.
- Uga, Y., K. Sugimoto, S. Ogawa, J. Rane, M. Ishitani, N. Hara, Y. Kitomi, Y. Inukai, K. Ono, N. Kanno et al. (2013) Control of root system architecture by DEEPER ROOTING 1 increases rice yield under drought conditions. Nat Genet 45: 1097–1102.
- Uga, Y., Y. Kitomi, S. Ishikawa and M. Yano (2015a) Genetic improvement for root growth angle to enhance crop production. Breed Sci 65: 111–119.
- Uga, Y., Y. Kitomi, E. Yamamoto, N. Kanno, S. Kawai, T. Mizubayashi and S. Fukuoka (2015b) A QTL for root growth angle on rice chromosome 7 is involved in the genetic pathway of DEEPER ROOTING 1. Rice (N Y) 8: 8.
- Wagih, O. (2017) ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics 33: 3645–3647.
- Wang, H., J. Wei, P. Li, Y. Wang, Z. Ge, J. Qian, Y. Fan, J. Ni, Y. Xu, Z. Yang et al. (2019a) Integrating GWAS and gene expression analysis identifies candidate genes for root morphology traits in Maize at the seedling stage. Genes (Basel) 10: 773.
- Wang, J., X. Liu, A. Zhang, Y. Ren, F. Wu, G. Wang, Y. Xu, C. Lei, S. Zhu, T. Pan et al. (2019b) A cyclic nucleotide-gated channel mediates cytoplasmic calcium elevation and disease resistance in rice. Cell Res 29: 820–831.
- Wang, K.L.C., H. Yoshida, C. Lurin and J.R. Ecker (2004) Regulation of ethylene gas biosynthesis by the Arabidopsis ETO1 protein. Nature 428: 945–950.
- Xu, Y., J. Yang, Y. Wang, J. Wang, Y. Yu, Y. Long, Y. Wang, H. Zhang, Y. Ren, J. Chen et al. (2017) OsCNGC13 promotes seed-setting rate by facilitating pollen tube growth in stylar tissues. PLoS Genet 13: 7.
- Yang, J.-C., H. Zhang and J.-H. Zhang (2012) Root morphology and physiology in relation to the yield formation of rice. J Integr Agric 11: 920–926.
- Yano, K., E. Yamamoto, K. Aya, H. Takeuchi, P.C. Lo, L. Hu, M. Yamasaki, S. Yoshida, H. Kitano, K. Hirano et al. (2016) Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat Genet 48: 927–934.
- Yokoo, M., F. Kikuchi, A. Nakane and H. Fujimaki (1980) Genetical analysis of heading time by aid of close linkage with blast resistance in rice. Bull Natl Inst Agr Sci D 31: 95–126.
- Yonemaru, J., T. Yamamoto, S. Fukuoka, Y. Uga, K. Hori and M. Yano (2010) Q-TARO: QTL annotation rice online database. Rice (N Y) 3: 194–203.
- Yoshida, S., D.A. Forno, J.H. Cock and others (1976) Laboratory manual for physiological studies of rice. Third edition. International Rice Research Institute, Los Banos, Philippines.
- Yu, J., G. Pressoir, W.H. Briggs, I.V. Bi, M. Yamasaki, J.F. Doebley, M.D. McMullen, B.S. Gaut, D.M. Nielsen, J.B. Holland et al. (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38: 203–208.
- Zhang, W.P., X.Y. Shen, P. Wu, B. Hu and C.Y. Liao (2001) QTLs and epistasis for seminal root length under a different water supply in rice (Oryza sativa L.). Theor Appl Genet 103: 118–123.
- Zhao, Z., Y. Zhang, X. Liu, X. Zhang, S. Liu, X. Yu, Y. Ren, X. Zheng, K. Zhou, L. Jiang et al. (2013) A role for a dioxygenase in auxin metabolism and reproductive development in rice. Dev Cell 27: 113–122.