Abstract
The isolation of disease resistance genes introduced from wild or related cultivated species is essential for understanding their mechanisms, spectrum and risk of breakdown. To identify target genes not included in reference genomes, genomic sequences with the target locus must be reconstructed. However, de novo assembly approaches of the entire genome, such as those used for constructing reference genomes, are complicated in higher plants. Moreover, in the autotetraploid potato, the heterozygous regions and repetitive structures located around disease resistance gene clusters fragment the genomes into short contigs, making it challenging to identify resistance genes. In this study, we report that a de novo assembly approach of a target gene-specific homozygous dihaploid developed through haploid induction was suitable for gene isolation in potatoes using the potato virus Y resistance gene Rychc as a model. The assembled contig containing Rychc-linked markers was 3.3 Mb in length and could be joined with gene location information from the fine mapping analysis. Rychc was successfully identified in a repeated island located on the distal end of the long arm of chromosome 9 as a Toll/interleukin-1 receptor-nucleotide-binding site-leucine rich repeat (TIR-NBS-LRR) type resistance gene. This approach will be practical for other gene isolation projects in potatoes.
Introduction
The isolation of the causal genes responsible for disease resistance in crops is not only an essential study for elucidating the disease resistance mechanism but is also important for evaluating its utility in breeding, such as estimating resistance intensity and spectrum and the risk of resistance breakdown. In potato (Solanum tuberosum L.) breeding programs, many resistance genes against late blight (Paluchowska et al. 2022), potato cyst nematode (Gartner et al. 2021), potato virus Y (PVY) (Grech-Baran et al. 2020, Nie et al. 2016, Szajko et al. 2014), potato virus X (PVX) (Ritter et al. 1991, Van der Vossen et al. 2000), bacterial wilt caused by Ralstonia solanacearum (Habe et al. 2019, Mori et al. 2012) have been introduced, but some of resistance genes are still not cloned.
In recent years, several methods used to obtain high-density genotypes linked to phenotypes have become more accessible by combining high-quality reference genomes with next-generation sequencing (NGS)-based techniques, such as double-digest restriction site-associated DNA sequencing (ddRAD-seq) (Shirasawa et al. 2016), Genotyping by random amplicon sequencing-direct (GRAS-Di) (Enoki and Takeuchi 2018, Hosoya et al. 2019), MutMap+ (Fekih et al. 2013), and QTL-seq (Takagi et al. 2013, Yamakawa et al. 2021). These technologies have enabled us to obtain the locus positions of target genes and information on linked DNA markers for fine mapping at high speed and with high accuracy. However, regardless of how much candidate regions are narrowed down by fine mapping, isolating disease resistance genes derived from wild or cultivated relatives that are distantly related to the reference genome remains challenging. This is because the reference genomes do not contain these genes, the resistance genes are often clustered, and the region involves structural variations associated with gene duplications. In such cases, the sequence of the target gene locus must be reconstructed newly.
There are two significant issues in the isolation of target genes via a de novo assembly approach in polyploid crops. First, genome assemblers cannot reconstruct the heterozygous regions into one contiguous sequence because the assemblers fragment contigs to preserve information on haplotypes (Garg et al. 2018, Kajitani et al. 2014, 2019, Schatz et al. 2012). To avoid heterozygosity, most of the previously reported reference genomes of potatoes were reconstructed from pure lines or doubled monoploids (Pham et al. 2020, Van Lieshout et al. 2020, Xu et al. 2011). Second, the repetitive structure caused by gene duplication events or translocation events of transposable elements also causes fragmentation of contigs (Alkan et al. 2011, Schatz et al. 2012). For these reasons, if the same approach as that used in the previous reference genome construction projects was selected for gene isolation, the cost of creating materials, sequencing, and computer analysis would be high. Particularly, the autotetraploid potato has highly heterozygous genomes; therefore, it is nearly impossible to assemble contigs that are sufficiently long for map-based cloning.
However, when information about the target gene locus is available, chromosome-scale genome assembly is unnecessary for gene isolation (Grech-Baran et al. 2020). If we can create lines homozygous in the target region, we should be able to obtain the sequence of the target locus with high accuracy using the de novo genome assembly approach. In the case of potatoes, homozygous dihaploid lines (RR or rr) could be obtained from dihaploid induction (Busse et al. 2021, Hutten et al. 1993, Peloquin et al. 1996, Van Breukelen et al. 1977) of duplex tetraploid lines (RRrr) with a probability of 1/6. On the other hand, homozygous tetraploid lines (RRRR or rrrr) could be obtained with a probability of 1/36 from crosses of duplex tetraploid RRrr × RRrr. Therefore, it is easier to obtain the homozygous line by the dihaploid induction approach than crossing in the tetraploid level. Also, in the allele dosage estimation analysis by quantitive PCR or progeny tests, homozygosity is easier to determine in dihaploid than in tetraploids. Such target gene specific homozygous dihaploid lines would be unsuitable for de novo assembly of the entire genome because most of the genome is heterozygous, but they are helpful for the assembly aimed at targeted gene isolation.
Moreover, third-generation sequencing (TGS) is effective in solving repetitive structures. The nanopore sequencing technology offered by Oxford Nanopore Technology and the single molecule real-time (SMRT) sequencing technology offered by Pacific Biosciences can output very long reads (>15 kb). These long reads can overcome the scattered repeats in the genome, thus facilitating the resolution of graph structures during de novo assembly and significantly improving the continuity of contigs (Kono and Arakawa 2019).
Rychc derived from an wild potato (S. chacoense) is a valuable gene for potato breeding programs, as it confers extreme resistance against PVY infection. Rychc was first introduced into the commercial cultivar ‘Konafubuki’ from doubled S. chacoense ‘w84’(Asama et al. 1982) and was mapped to the distal end of the long arm of chromosome 9 (Hosaka et al. 2001, Sato et al. 2006, Takeuchi et al. 2008). Moreover, Li et al. (2022) reported the cloning of the Rychc gene as a Toll/interleukin-1 receptor (TIR)-nucleotide-binding site (NBS)-leucine rich repeat (LRR) type resistance gene by screening bacterial artificial chromosome (BAC) clones.
In this study, we report that the de novo genome assembly of a partial homozygous dihaploid line by TGS is a more accessible and accurate approach for gene isolation in potatoes, rather than the high-cost BAC/Yeast artificial chromosome (YAC) cloning systems. We identified the Rychc gene as a model and revealed that it is located in the repetitive structure at the distal end of the long arm of chromosome 9.
Materials and Methods
Fine mapping analysis
Recombinants between two markers, RY364 and RY186 (Mori et al. 2011, Takeuchi et al. 2008), were screened from eight populations derived from crosses between Rychc+ and Rychc– tetraploid lines/varieties. Supplemental Table 1 lists the population name, cross combination, and the number of individuals in each population. The progeny lines used in this research were named ‘population name – individual number’.
Seeds were sown on nursery beds, and genomic DNA was extracted from leaf tips using the TPS buffer (100 mM Tris-HCl (pH 8.0), 1 M KCl, and 10 mM EDTA) (Hattori et al. 2007). Polymerase chain reaction (PCR) was performed according to Takeuchi et al. (2008). Recombinants were transplanted to 9 cm vinyl pots and cultivated until harvesting. DNA markers in the candidate region were designed based on the genomic sequence of S. phureja DM1-3 ver4.03 obtained from Spud DB (http://spuddb.uga.edu/index.shtml). Primer pairs were designed to amplify the intergenic or intronic region, and its band pattern was compared between ‘Konafubuki’ (Rychc+) and ‘Hokkaikogane’ (Rychc–) cultivars. The primers that exhibited polymorphism were used for fine mapping (Supplemental Table 2).
The total volume in the PCR assay was 10 μl, comprising 2 μl of template DNA, 5 μl of gene amplification reagent (Ampdirect Plus®, Shimadzu Corp., Kyoto, Japan), 0.25 U Taq DNA polymerase (BIOTAQ HSTM; Bioline, London, UK), and the corresponding primer pair. Thermal cycling was performed using 96- or 384-well thermal cyclers (Veriti, Applied Biosystems, Life Technologies, Carlsbad, CA, USA). The PCR conditions for RY122 (RY122-18/RY122-19), RY173 (RY173-10/RY173-27), RY186 (RY186-11/RY186-12), RY364 (Ry364-14/Ry364-19), ASRY22 (ASRY22-F/ASRY22-R), ASRY49 (ASRY49-F/ASRY49-R), ASRY63 (ASRY63-F/ASRY63-R) ASRY91 (ASRY91-F/ASRY91-R) and ASRY76 (ASRY76-F/ASRY76-R) consisted of one cycle of 10 min at 94°C; followed by 35 cycles of 30 s at 94°C, 30 s at 55°C, and 1 min at 72°C; and one final cycle of 5 min at 72°C. The PCR conditions for SZRY54 (SZRY54-5b/SZRY54-5g), SZRY183 (SZRY183-4m/SZRY183-4n), SZRY50 (SZRY50-2d/SZRY50-3f), and SZRY119 (SZRY119-F/SZRY119-R) consisted of one cycle of 10 min at 94°C; followed by 35 cycles of 30 s at 94°C, 30 s at 60°C, and 5 min at 72°C; and one final cycle of 5 min at 72°C. The PCR conditions for SZRY93 (SZRY93-4j/SZRY93-6k) consisted of one cycle of 10 min at 94°C; followed by 35 cycles of 30 s at 94°C, 30 s at 60°C, and 2 min at 72°C; and one final cycle of 5 min at 72°C. All PCR products were separated via electrophoresis on 2.0%–3.0% agarose gels in 1× TAE buffer (40 mM Tirs, 20 mM acetic acid, and 1 mM EDTA) and visualized through the SYBR Safe DNA gel stain (Invitrogen, Life Technologies, Carlsbad, CA, USA) and UV transillumination.
Inoculation of PVY
For mechanical inoculation, a virus inoculum was prepared by grinding the infected leaves of Nicotiana tabacum ‘Xanthi nc’ in 10 volumes of 50 mM potassium phosphate buffer (pH 7.0) including 0.1% (w/v) sodium sulfite. The fifth and sixth leaves of potato plants at the 8–9 leaf stage (approximately 4 weeks after budding) were dusted with carborundum and mechanically inoculated using a swab. The plants were grown in a growth chamber at 22°C with 16 h of light (10,000 lux). To confirm systemic infection, the top two leaves (noninoculated upper leaves) were sampled at 21 dpi and then assayed for PVY via Triple antibody sandwich enzyme-linked immune sorbent assay (TAS-ELISA) using the ELISA Reagent Set for Potato virus Y (Agdia, Elkhar, IN, USA). Samples showing absorbance values greater than twice that of the noninoculated plants were judged to be susceptible to PVY.
Development of a potato line for genome sequencing
We crossed the pollen of the haploid inducer S. phureja ‘IVP35’ to Rychc duplex tetraploid lines ‘13082-3’ (‘Saikai 37’ × ‘Konafubuki’) and 13077-7’ (‘Saikai 35’ × ‘Hokuiku 20’). The ploidy of the progenies was analyzed using a CyFlow Ploidy Analyzer (Sysmex, Kobe, Japan), and dihaploid lines were established. Continuously, the DNA markers RY186 and RY364 were amplified via PCR. Allele dosage at the Rychc locus was estimated from the ratio of amplification of RY364 against the internal control aprt in quantitative PCR from gDNA, as described by Asano and Tamiya (2013). The dihaploid ‘98H20-5’ was used as a control with heterozygous Rychc. DNA from each progeny was extracted using DNeasy plant mini kits (QIAGEN, Venlo, Netherland). The total volume in the qPCR assay was 20 μl, comprising 2 μl of template DNA (20 ng/μl), 10 μl of SYBR Green Master Mix (Thermo Fisher Scientific, Waltham, MA, USA), and 0.1 μM forward and reverse specific primers.
Whole-genome sequencing
For Nanopore and Illumina whole-genome sequencing, high-molecular-weight DNA was extracted from 1 g of young leaves of the line ‘184202-2’ using a Nucleobond HMW DNA kit (MACHEREY-NAGEL, Düren, Germany). We followed the manufacturer’s protocol for extraction, except for the lysing step, which was extended to 3 h. Short DNA fragments (<15 kbp) were removed from total DNA using a Short Read Eliminator (Circulomics, Pacbio, Menlo Park, CA, USA) and resuspended in TE buffer (pH 8.0). We prepared a nanopore sequencing library using SQK-LSK109 (Oxford Nanopore technology, Oxford, UK) and sequenced it three times with two FLO-MIN106D flow cells on a MinION sequencer. FAST5 data were base called using Guppy ver3.4.5 in the high accuracy mode with NVIDIA GPU (GTX1060 3GB). The adapter sequence was trimmed using the Porechop software ver0.12 (https://github.com/rrwick/Porechop). Low-quality reads (mean quality score (QS) <8) and short reads (<3000 bp) were removed using NanoFilt (De Coster et al. 2018). Subsequently, chimeric reads were split using yacrd (Marijon et al. 2020). Illumina whole-genome sequencing (150 bp paired-end) was performed on an Illumina Hiseq X Ten sequencer with Hiseq X Ten Reagent Kit v2.5 (outsourced to Eurofins Genomics). For preprocessing Illumina reads, low-quality reads (mean QS <30) and short reads (<50 bp) were filtered out using Fastp ver0.20.0 (Chen et al. 2018).
De novo assembly
Nanopore long reads were de novo assembled using Flye ver2.7 (Kolmogorov et al. 2019) with the --nano-raw option. The obtained contigs were subjected to five runs of the Racon consensus caller ver1.4.13 (Vaser et al. 2017) using long reads to obtain a consensus sequence, and chimeric contigs were split using Medaka ver1.1.2 (Oxford Nanopore Technologies). Errors were further corrected three times using Pilon ver1.23 (Walker et al. 2014) using Illumina short read data. The assembled results were evaluated on the solanales_odb10 dataset using BUSCO ver5.3.2 (Simão et al. 2015). The sequences of DNA markers were searched on the contigs using NCBI-blast+ ver2.10.0 (Zhang et al. 2000). A dot plot analysis was conducted using D-GENIES (Cabanettes and Klopp 2018) and Gepard ver1.20 (Krumsiek et al. 2007).
RNA-seq and gene prediction
Total RNA was extracted from fully expanded leaves of dihaploid ‘184202-2’ 30 days after budding using the Promega Maxwell RSC Plant RNA kit (Promega, Madison, WI, USA). Libraries were created from this RNA using the TruSeq Stranded mRNA Library Prep Kit and sequenced at 150 bp paired-end using NovaSeq6000 (outsourced to Eurofins Genomics). RNA-seq reads were extracted using Fastp for reads with QS >30 and mapped to contigs obtained via the de novo genome assembly of ‘184202-2’ using HISAT2.2.1 (Kim et al. 2019). The output of HISAT2 was converted to BAM files using Samtools ver1.9 (Li et al. 2009), and StringTie2 ver2.1.2 (Kovaka et al. 2019) was used for mRNA assembly and gene prediction. Additionally, predicted gene sequences were obtained from the GTF files using Gffread (Pertea and Pertea 2020). Transcripts found within the candidate regions were subjected to a Gene ontology analysis and BLAST search in OmicsBox (BioBam, Spain), to infer their functions.
Cloning of candidate genes
Specific primers were manually designed in the flanking region of eight candidate genes obtained by StringTie2. The total volume of each PCR reaction was 20 μl, including 150 ng of total DNA, 10 μl of KOD One Master mix (TOYOBO, Japan), and 1 μM specific primers. PCR amplification conditions were 30 cycles of 98°C for 10 s, 65°C for 5 s, and 68°C for 1 min using an ABI9700 thermal cycler (Applied Biosystems, USA). The amplified fragments were ligated into the pCR-BluntII-TOPO vector (Thermo Fisher Scientific) and transformed into Escherichia coli DH5α cells. Each sublined vector was sequenced by Sanger sequencing using BigDye terminator ver3.1 (Thermo Fisher Scientific) with candidate genes specific primers shown in Supplemental Table 2.
Data availability
The whole-genome sequencing and RNA-seq data have been deposited in the DNA Data Bank of Japan (DDBJ) repository under project number PRJDB13325. The accession numbers of each sequencing data are below. The accession numbers of nanopore whole-genome sequencing data are DRR402764, DRR402765, and DRR402766. The accession number of whole-genome sequencing data by Illumina Hiseq X pair-end is DRR402767. The accession number of mRNA sequencing data by Illumina Novaseq6000 pair-end is DRR402768. The genomic sequence of Rychc detected in this research has been deposited as LC726345 to DDBJ.
Results
Fine mapping of Rychc
Before developing a Rychc homozygous line, fine mapping was performed to narrow down the candidate region of Rychc. In total, 62 recombinants between RY186 and RY364 were screened out from 21,364 F1 plants generated by eight crosses between Rychc+ and Rychc– lines/varieties (Supplemental Table 1). Each recombinant was genotyped using 12 newly developed DNA markers, and recombinants identified to be with recombination in the outer regions than inner DNA markers were excluded from further analysis. As a result of genotyping and phenotyping, the nearest recombinants from the population ‘13099’ obtained from the cross between ‘Sakurafubuki (Rychc+)’ and ‘Hokkaikogane (Rychc–)’. One recombinant (‘13099-2’) was identified between Rychc and SZRY54; on the other side of Rychc, three recombinants (‘13099-8’, ‘13099-17’, and ‘13099-23’) were identified between Rychc and marker SZRY183. As a result of the fine mapping, Rychc was delimited between SZRY183 and SZRY54 (Fig. 1, Supplemental Table 3).

To reduce the de novo genome assembly difficulties caused by the heterozygous genome of autotetraploid potato, we developed a dihaploid line with homozygous Rychc from ‘13077-7’ and ‘13082-3’, which carry duplex Rychc. From 16 and 17 berries, respectively, we obtained 14 and 22 true seeds without embryo spots, which are indicators of triploid or tetraploid hybrids between tetraploid and ‘IVP35’ (Hutten et al. 1993). We generated nine and seven dihaploid individuals, respectively, nine and six of which were determined to have Rychc from the results of the PCR amplification of RY364 and RY186 (Supplemental Table 4). Quantitative PCR revealed that only one line, named ‘184202-2’, obtained from the cross between ‘13082-3’ × ‘IVP35’, showed twice as much amplification of RY364 as the Rychc heterozygous dihaploid line ‘98H20-5’ (data not shown). Moreover, progeny tests from a cross between the diploid cultivar ‘Inka-no-mezame’ (Rychc–), which dose not posses RY186 and RY364, and ‘184202-2’ showed that all individuals had RY364 and RY186 (data not shown). From these results, ‘184202-2’ is confirmed as a dihaploid line with a homozygous Rychc region.
Furthermore, ‘184202-2’ produced tubers, and had female and male fertility in the greenhouse condition (25°C–35°C) without necrosis, aerial tubers, dwarfing, or early growth arrest. It showed extreme resistance to PVYO and PVYNTN; however, under high-temperature conditions (28°C) in the growth chamber, ‘184202-2’ formed hypersensitive reaction lesions against PVY inoculation, similar to the other varieties with Rychc (‘Konafubuki’ and ‘Sakurafubki’) described by Ohki et al. (2018) (data not shown). Therefore, ‘184202-2’ had all the characteristics of Rychc and could be used for further analysis.
De novo genome assembly and gene annotation
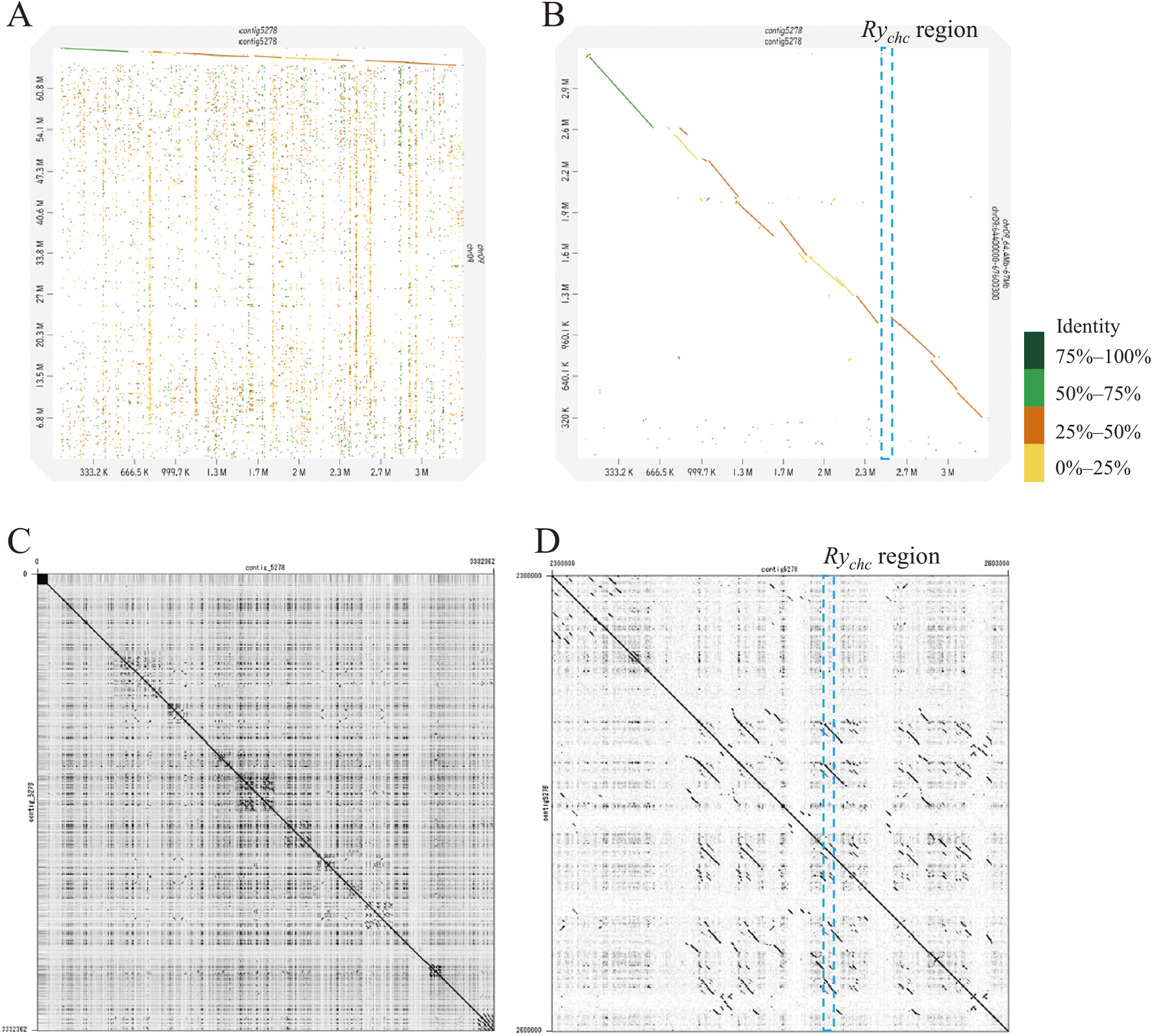
Table 1 shows the statistics of NGS and TGS data used in this study. De novo assembly by Flye from about 12 Gb of nanopore long reads generated 9,622 contigs, with the largest contig being 9.67 Mb long and contig N50 being 352 kb (Table 2). After error polishing using Pilon v1.23 with about 80× of Illumina short reads, the assembled contigs were subjected to a BUSCO ver. 5.3.2 analysis, to assess the completeness of the assembly. In the 5,950-gene set of the solanales_odb10 dataset, 96.9% of genes were identified (83.0% were single and 13.9% were duplicated), 0.9% were fragmented, and 2.2% were missing (Table 2). Compared with the reference genomes of potatoes, the assembly of ‘184202-2’ was considered suitable for searching the Rychc region. Also, Contig5278 was homologous to the distal end of the long arm of chromosome 9 of S. phureja DM1-3 v6.1 (Pham et al. 2020) (Fig. 2A). However, the sequence identity was under 50% around the Rychc region in Contig5278 (Fig. 2B). A self-dot plot analysis revealed a highly repeated structure for Contig5278 (Fig. 2C), and the Rychc region was located in a duplicated island (Fig. 2D). Next, we searched the primer sites of DNA markers linked to Rychc in the contigs through BLAST search. Among these contigs, only Contig5278 (3.3 Mb) included markers RY186 and RY364 linked to Rychc; RY186 and RY364 were contained in a region of approximately 635 kb (Fig. 3A). Moreover, sites of other DNA markers designed in the fine mapping were found between RY186 and RY364.
Table 1.
Statistics of the genome sequencing data used for
de novo assembly and RNA-seq data
| Nanopore WGS |
Total bases (bp) |
12,900,979,133 |
|
Total reads |
725,512 |
|
Average read length (bp) |
17,781.9 |
|
Longest read length (bp) |
250,644 |
|
Mean QS |
11.5 |
|
Read N50 (bp) |
34,181 |
| Illumina WGS |
Total bases (bp) |
82,236,131,442 |
|
Total reads |
544,610,142 |
|
Read length (bp) |
150 |
|
QS >30 |
93.6% |
|
QS >20 |
97.2% |
| Illumina RNA-seq |
Total bases (bp) |
8,412,300,300 |
|
Total reads |
56,082,002 |
|
Read length (bp) |
150 |
|
QS >30 |
94.3% |
|
QS >20 |
97.6% |
Table 2.
Statistics of the genome assembly of ‘184202-2’
|
Genome assembly |
| S. tuberosum 184202-2 |
S. phureja DM1-3 v4.04 |
S. phureja DM1-3 v6.01 |
S. chacoense M6 v4.1 |
| Parameters |
(this study) |
(Hardigan et al. 2016) |
(Pham et al. 2020) |
(Leisner et al. 2018) |
| Methods |
Nanopore long reads and Illumina |
Illumina and Sanger |
Nanopore long reads |
Illumina |
| Total assembly size (Mb) |
1,091.1 |
884.1 |
741.6 |
825.7 |
| Number of contigs |
9,622 |
170,833 |
1,382 |
– |
| Number of scaffolds |
– |
14,853 |
288 |
8,260 |
| Largest contig length (bp) |
9,678,383 |
– |
– |
– |
| Average contig length (bp) |
113,393 |
– |
– |
– |
| Largest scaffold length (bp) |
– |
111,078,864 |
88,591,686 |
7,385,816 |
| Average scaffold length (bp) |
– |
63,150,592 |
2,574,948 |
99,972 |
| Contig N50 (bp) |
352,213 |
170,833 |
17,312,182 |
– |
| Scaffold N50 (bp) |
– |
1,344,915 |
59,670,755 |
713,601 |
| BUSCO ver5.3.2 with solanales_odb10 dataset |
|
|
|
|
| Total |
5,950 |
5,950 |
5,950 |
5,950 |
| Complete |
96.9% |
98.6% |
98.7% |
89.8% |
| single |
83.0% |
96.3% |
96.4% |
87.5% |
| duplicated |
13.9% |
2.3% |
2.3% |
2.3% |
| Fragmented |
0.9% |
0.2% |
0.1% |
0.2% |
| Missing |
2.2% |
1.2% |
1.2% |
10.0% |

Rychc induces resistance against PVY inoculation on leaves, which should be detected via gene expression analysis in leaves. RNA-seq analysis from matured leaves of ‘184202-2’ predicted 27 genes between the 2.3–2.6 Mb point and the Rychc candidate region (Fig. 3B). “STRG” is the default prefix of StringTie2. A BLAST analysis led to the annotation of eight of these 27 genes as Rpi-vnt1-like (CC-NBS-LRR) or tobacco mosaic virus (TMV) resistance gene N-like (TIR-NBS-LRR)-type resistance genes (Table 3). However, six of the eight resistance candidate genes had early stop codons in the sequence and encoded truncated proteins; therefore, these genes were not likely to be the Rychc gene. The genome sequences of each candidate genes were shown in Supplemental Table 5. Furthermore, based on the locus position of Rychc, as inferred based on fine mapping and the structures of these candidate genes, STRG1648 was expected to be Rychc.
Table 3.
Predicted genes in the
Rychc region and their properties
| Gene Name |
BLAST Description |
Start position on Contig5278 (bp) |
Gene length (nt) |
Transcript length (nt) |
Protein length (aa) from Kozak sequence |
Gene type |
| STRG1629 |
60S acidic ribosomal protein P0 |
2,292,859 |
1,006 |
1,006 |
280 |
|
| STRG1627 |
---NA--- |
2,298,771 |
911 |
911 |
1 |
|
| STRG1631 |
Rpi-vnt1-like protein |
2,300,618 |
2,895 |
2,895 |
864 |
CC-NBS-LRR |
| STRG1630 |
Late blight resistance protein, putative |
2,307,144 |
297 |
297 |
2 |
|
| STRG1632 |
Rpi-vnt1-like protein |
2,310,970 |
1,783 |
1,783 |
20 |
|
| STRG1633 |
Hypothetical protein SDM1_42t00007 |
2,316,039 |
210 |
210 |
No ATG found |
|
| STRG1634 |
---NA--- |
2,321,599 |
1,326 |
557 |
3 |
|
| STRG1635 |
Rpi-vnt1-like protein |
2,324,216 |
3,016 |
2,917 |
664 |
NBS-LRR |
| STRG1636 |
Protein ECERIFERUM 26-like |
2,348,256 |
1,080 |
1,080 |
21 |
|
| STRG1637 |
Chaperone protein dnaj 20, chloroplastic-like |
2,358,580 |
1,840 |
737 |
2 |
|
| STRG1642 |
Serine/arginine repetitive matrix protein 2 |
2,365,985 |
8,369 |
4,138 |
900 |
|
|
|
|
|
4,435 |
900 |
|
|
|
|
|
4,054 |
900 |
|
|
|
|
|
3,669 |
900 |
|
|
|
|
|
3,694 |
900 |
|
|
|
|
|
3,665 |
900 |
|
| STRG1638 |
Small subunit processome component 20 homolog |
2,382,753 |
1,154 |
734 |
2 |
|
| STRG1639 |
Small subunit processome component 20 homolog |
2,389,043 |
2,096 |
1,435 |
405 |
|
| STRG1640 |
Small subunit processome component 20 homolog |
2,391,240 |
1,084 |
996 |
330 |
|
| STRG1641 |
Small subunit processome component 20 homolog |
2,393,727 |
369 |
369 |
61 |
|
|
|
|
|
1,189 |
258 |
|
| STRG1643 |
TMV resistance protein N-like |
2,397,081 |
2,677 |
2,677 |
49 |
LRR |
| STRG1644 |
Deacetoxyvindoline 4-hydroxylase-like |
2,407,359 |
942 |
456 |
143 |
|
| STRG1645 |
Small subunit processome component 20 homolog |
2,418,710 |
816 |
816 |
10 |
|
| STRG1646 |
TMV resistance protein N-like |
2,423,296 |
2,043 |
2,043 |
49 |
LRR |
| STRG1648 |
TMV resistance protein N-like |
2,476,328 |
4,294 |
3,593 |
911 |
TIR-NBS-LRR |
|
|
|
|
4,047 |
554 |
TIR-NBS-WingedHelix |
| STRG1647 |
---NA--- |
2,502,697 |
348 |
348 |
22 |
|
| STRG1651 |
Small subunit processome component 20 homolog |
2,541,640 |
4,981 |
3,070 |
200 |
|
|
|
|
|
2,523 |
200 |
|
| STRG1649 |
TMV resistance protein N-like |
2,556,562 |
2,067 |
1,820 |
552 |
TIR-NBS-WingedHelix |
| STRG1650 |
TMV resistance protein N-like |
2,565,200 |
2,003 |
1,914 |
373 |
partial LRR |
| STRG1652 |
Protein RALF-like 32 |
2,589,214 |
467 |
467 |
8 |
|
| STRG1653 |
Probable magnesium transporter NIPA2 |
2,597,949 |
603 |
360 |
80 |
|
| STRG1654 |
Stress response protein NST1-like |
2,600,362 |
2,366 |
2,366 |
48 |
|
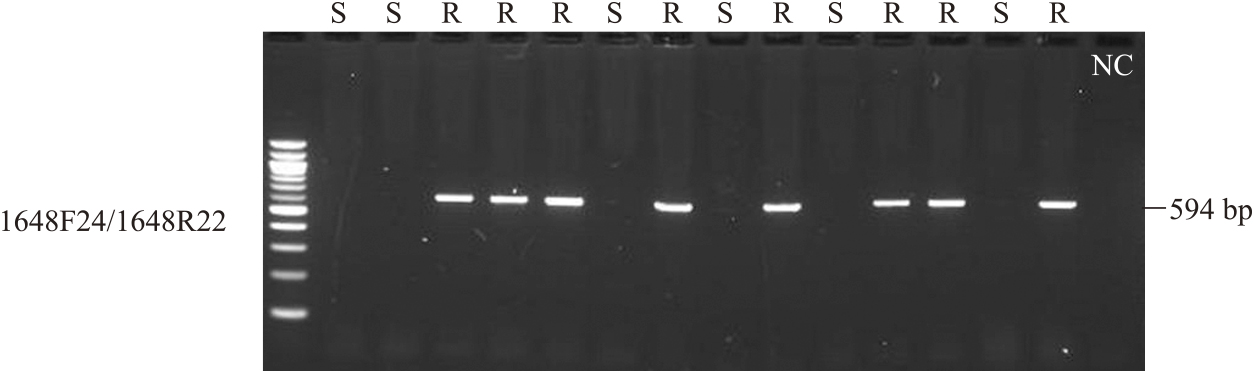
An RNA-seq analysis showed that STRG1648 had two splicing variants (Fig. 3C). The STRG1648.1 variant was predicted to have three introns in the transcript, encoding a 911-amino acid TIR-NBS-LRR-type protein (Table 3). In turn, the STRG1648.2 variant was predicted to skip the second and third introns and had a stop codon at the start of the LRR region; thus, the STRG1648.2 variant encoded a truncated Winged-Helix domain, instead of the LRR domain (Fig. 3C). Additionally, the presence/absence of STRG1648 amplification via PCR (1648F24/1648R22) correlated perfectly with the PVY-resistant phenotype (Fig. 4, Supplemental Fig. 3, Supplemental Table 3).
Nucleotide accuracy of the de novo assembly
The sequence of STRG1648 obtained from the de novo assembly perfectly matched the sequence of the PCR amplicon obtained using the primer pair STRG1648f-F/STRG1648f-R, as determined via Sanger sequencing (Supplemental Fig. 1). This result indicates that the nucleotide determination accuracy after polishing by Illumina reads was very high. Moreover, Supplemental Fig. 1 shows the comparison of the genome sequence of the STRG1648 gene with the Rychc sequence reported by Li et al. (2022). The nucleotide identity between Rychc and STRG1648 was 98% (4108/4176 bp) for the entire gene; therefore, we concluded that STRG1648 was the Rychc gene. Additionally, single nucleotide polymorphisms, insertions, and deletions were detected between two Rychc sequences (Supplemental Fig. 1).
Discussion
Long-read-based de novo assembly of the Rychc homozygous dihaploid
The Rychc region was estimated to be located in the NBS-LRR cluster with many repeat sequences because the reference genome DM1-3 ver4.03 (Xu et al. 2011), which was reconstructed by Sanger and Illumina sequencing, contains many assembly gaps in the Rychc region. To assemble contigs that are sufficiently long to combine the results from the fine mapping analysis, we chose de novo assembling using nanopore sequencing technology and error polishing with Illumina high-quality short reads. Moreover, the Rychc homozygous line ‘184202-2’ was developed using the dihaploid induction technique for genome sequencing.
As a result of the de novo assembly using nanopore long reads and error polishing by Illumina reads, we generated a 3.3-Mb-long contig (Contig5278) of this challenging region (Fig. 3). In Contig5278, one TIR-NBS-LRR-type resistance gene, named STRG1648, was identified between the closest markers, SZRY183 and SZRY54. Its sequence exhibited high homology with that of Rychc reported by Li et al. (2022), and the presence/absence of STRG1648 was perfectly correlated with the PVY resistance phenotype; therefore, we concluded that STRG1648 was the Rychc gene derived from S. chacoense ‘w84’. This strategy provided sufficient results to combine with the fine mapping results and showed that it is not necessary to construct BAC clones for map-based cloning in potatoes. Moreover, after error correction using short reads, the assembled nucleotide sequences of the Rychc gene (STRG1648) matched perfectly with the sequences of its PCR product, as determined by Sanger sequencing. This result implies that the contigs obtained in this study were sufficiently accurate for searching not only SCAR markers but also SNP or SSR markers.
Furthermore, in recent years, long-read sequencing technologies with base-call accuracies of 99% or better have also emerged, thus solving highly heterozygous genomes, such as the RH line of diploid potato (Zhou et al. 2020), the commercial tetraploid cultivar ‘Otava’ (Sun et al. 2022), and 44 diploid potato wild species (Tang et al. 2022). However, such haplotype-aware assemblies still require a multi-steps assembling approach containing genetic mapping on a sequenced F2 population, 20x–40x of the accurate long reads of Circular Consensus Sequencing or HiFi sequencing (Pacific Biosciences) and combinations of several technologies, such as Hi-C or Bionano (Zhou et al. 2020). From the point of view of cost, our approach with low cost Nanopore long reads is a simpler way to obtain target sequences.
Rychc is a TIR-NBS-LRR-type resistance gene
Rychc (STRG1648) was predicted to encode a protein consisting of 911 amino acid residues of the TIR-NBS-LRR type, similar to that reported by Li et al. (2022). Additionally, between two Rychc sequences, some nucleotide variations were detected; therefore, these two Rychc genes are likely in a homologous gene with similar functions that are maintained and are considered to be within the Rychc gene family.
In this study, we detected two splicing variants of Rychc, one of which had second and third introns (Fig. 3C). For resistance against the TMV induced by the N gene derived from N. glutinosa, splicing variants of N transcripts containing first and second introns are required because these splicing variants enhance the expression of the N gene and regulate N protein accumulation (Ikeda et al. 2021). Two splicing variants predicted for Rychc may be required for the stable and robust resistance afforded by Rychc by playing a role in controlling Rychc expression and protein accumulation levels during PVY infection as the N gene.
PVY accumulates mutations and recombinations in its genome continuously (Ogawa et al. 2008, 2012, Rodriguez-Rodriguez et al. 2020); therefore, the possibility of PVY isolates overcoming Rychc resistance cannot be ruled out. To evaluate this possibility, further studies are warranted to clarify in detail which proteins of PVY are recognized by Rychc as Avrs and what resistance mechanisms are at work.
The origin of Rychc
The number of NBS-LRR-type disease resistance genes has generally increased because of events such as large-scale tandem repeats caused by unequal crossing over. The newly duplicated genes are relieved of selection pressure and accumulate mutations, resulting in the recognition of various pathogens(Han and Tsuda 2022). The highly repeated sequence located around Rychc in Contig5278 suggests that many tandem duplication events occurred in the distal end of the long arm of chromosome 9 (Fig. 2). Moreover, a sequence comparison between Contig5278 and the reference genomes of S. chacoense ‘M6’ (Leisner et al. 2018) and S. tuberosum ‘Solyntus’ (Van Lieshout et al. 2020) showed the presence of various structural variants, such as inversions, duplications, and deletions (Supplemental Fig. 2).
Conversely, the accumulation of random mutations on duplicated resistance genes may induce an autoimmune response by recognizing the proteins they produce, which in turn trigger not only programmed cell death but also a systemic hypersensitive reaction. In such cases, mutations in the promoter region or some early stop codons appear to inactivate harmful resistance genes (Barragan et al. 2021). The fact that six of the eight disease resistance genes annotated in the Rychc locus have early stop codons suggests the existence of such natural selection.
Li et al. (2022) suggested that Rychc may have rapidly accumulated mutaions and obtained the ability to recognize PVY as the PVY geneome diversified and subsequently spread and maintained in the S. chacoense population. Traces of duplication events revealed in our study suggest that the region of Rychc has experienced multiple duplication events and may support the view of that the mutation expanded under the selection pressure of PVY described in Li et al. (2022). However, it is not possible to determine from these results when Rychc was emerged in the duplication events and when it acquired the ability of PVY recognition.
Conclusion
In this study, the PVY resistance gene Rychc was detected by map-based cloning using traditional haploid induction techniques and de novo genome assembly with long reads. Compared with the previously reported Rychc gene, the sequence of the Rychc identified in this study included several mutations, indicating that the Rychc family exhibits some diversity. The repeated sequences detected on Contig5278 were thought of as traces of the gene duplication events that led to the Rychc gene. Furthermore, the sequence of contigs obtained in this study was accurate, implying that any type of DNA marker can be detected. Also, further research on how Rychc recognizes PVY and initiates the resistance cascade will determine the future of Rychc use in PVY resistance potato breeding. Additionally, our gene isolation approach will also work well in the search for other valuable genes in potatoes.
Author Contribution Statement
K. Akai carried out the development of the dihaploid clone, whole-genome sequence, de novo assembly, and gene annotation. K. Asano, CS, TS, and TT designed primers for the fine mapping and screened recombinant lines. ES and ST carried out the development of Rychc duplex tetraploid lines. TO carried out PVY inoculation test and cloning of candidate genes. All authors were involved in improving this manuscript.
Acknowledgments
We thank Dr. Kazuyoshi Hosaka and Dr. Rena Sanetomo, Obihiro University of Agriculture and Veterinary Medicine, for providing a Ploidy Analyzer. We also thank Dr. Chikara Masuta, Research Faculty of Agriculture, Hokkaido University, for our discussions and advice on this study. The maintenance of the dihaploid line was supported by Mr. Satoshi Okamoto, Center for Seeds and Seedlings, NARO. This study was supported by grants from the Ministry of Agriculture, Forestry, and Fisheries of Japan (Genomics-Based Technology for Agricultural Improvement [SFC3002]).
Literature Cited
- Alkan, C., S. Sajjadian and E.E. Eichler (2011) Limitations of next-generation genome sequence assembly. Nat Methods 8: 61–65.
- Asama, K., H. Ito, N. Murakami and T. Ito (1982) New potato variety “Konafubuki”. Bulletin of Hokkaido Prefectural Agricultural Experiment Station 48: 75–84 (in Japanese with English summary).
- Asano, K. and S. Tamiya (2013) Development of rapid estimation method for allele number of H1 and selection of multiplex lines in potato. Breed Res 15 (Suppl. 1): 53 (in Japanese).
- Barragan, A.C., M. Collenberg, J. Wang, R.R.Q. Lee, W.Y. Cher, F.A. Rabanal, H. Ashkenazy, D. Weigel and E. Chae (2021) A truncated singleton NLR causes hybrid necrosis in Arabidopsis thaliana. Mol Biol Evol 38: 557–574.
- Busse, J.S., S.H. Jansky, H.I. Agha, C.A.S. Carley, L.M. Shannon and P.C. Bethke (2021) A high throughput method for generating dihaploids from tetraploid potato. Am J Potato Res 98: 304–314.
- Cabanettes, F. and C. Klopp (2018) D-GENIES: Dot plot large genomes in an interactive, efficient and simple way. PeerJ 6: e4958.
- Chen, S., Y. Zhou, Y. Chen and J. Gu (2018) Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34: i884–i890.
- De Coster, W., S. D’Hert, D.T. Schultz, M. Cruts and C. van Broeckhoven (2018) NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 34: 2666–2669.
- Enoki, H. and Y. Takeuchi (2018) New genotyping technology, GRAS-Di, using next generation sequencer. Proceedings of Plant and Animal Genome Conference XXVI.
- Fekih, R., H. Takagi, M. Tamiru, A. Abe, S. Natsume, H. Yaegashi, S. Sharma, S. Sharma, H. Kanzaki, H. Matsumura et al. (2013) MutMap+: Genetic mapping and mutant identification without crossing in rice. PLoS One 8: e0068529.
- Garg, S., M. Rautiainen, A.M. Novak, E. Garrison, R. Durbin and T. Marschall (2018) A graph-based approach to diploid genome assembly. Bioinformatics 34: i105–i114.
- Gartner, U., I. Hein, L.H. Brown, X. Chen, S. Mantelin, S.K. Sharma, L.M. Dandurand, J.C. Kuhl, J.T. Jones, G.J. Bryan et al. (2021) Resisting potato cyst nematodes with resistance. Front Plant Sci 12: 1–18.
- Grech-Baran, M., K. Witek, K. Szajko, A.I. Witek, K. Morgiewicz, I. Wasilewicz-Flis, H. Jakuczun, W. Marczewski, J.D.G. Jones and J. Hennig (2020) Extreme resistance to Potato virus Y in potato carrying the Rysto gene is mediated by a TIR-NLR immune receptor. Plant Biotechnol J 18: 655–667.
- Habe, I., K. Miyatake, T. Nunome, M. Yamasaki and T. Hayashi (2019) QTL analysis of resistance to bacterial wilt caused by Ralstonia solanacearum in potato. Breed Sci 69: 592–600.
- Han, X. and K. Tsuda (2022) Evolutionary footprint of plant immunity. Curr Opin Plant Biol 67: 102209.
- Hardigan, M.A., E. Crisovan, J.P. Hamilton, J. Kim, P. Laimbeer, C.P. Leisner, N.C. Manrique-Carpintero, L. Newton, G.M. Pham, B. Vailancourt et al. (2016) Genome reduction uncovers a large dispensable genome and adaptive role for copy number variation in asexually propagated Solanum tuberosum. Plant Cell 28: 388–405.
- Hattori, Y., K. Miura, K. Asano, E. Yamamoto, H. Mori, H. Kitano, M. Matsuoka and M. Ashikari (2007) A major QTL confers rapid internode elongation in response to water rise in deepwater rice. Breed Sci 57: 305–314.
- Hosaka, K., Y. Hosaka, M. Mori, T. Maida and H. Matsunaga (2001) Detection of a simplex RAPD marker linked to resistance to potato virus Y in a tetraploid potato. Am J Potato Res 78: 191–196.
- Hosoya, S., S. Hirase, K. Kikuchi, K. Nanjo, Y. Nakamura, H. Kohno and M. Sano (2019) Random PCR-based genotyping by sequencing technology GRAS-Di (genotyping by random amplicon sequencing, direct) reveals genetic structure of mangrove fishes. Mol Ecol Resour 19: 1153–1163.
- Hutten, R.C.B., E.J.M.M. Scholberg, D.J. Huigen, J.G.T. Hermsen and E. Jacobsen (1993) Analysis of dihaploid induction and production ability and seed parent × pollinator interaction in potato. Euphytica 72: 61–64.
- Ikeda, C., K. Taku, T. Miyazaki, R. Shirai, R.S. Nelson, H. Nyunoya, Y. Matsushita and N. Sasaki (2021) Cooperative roles of introns 1 and 2 of tobacco resistance gene N in enhanced N transcript expression and antiviral defense responses. Sci Rep 11: 15424.
- Kajitani, R., K. Toshimoto, H. Noguchi, A. Toyoda, Y. Ogura, M. Okuno, M. Yabana, M. Harada, E. Nahayasu, H. Maruyama et al. (2014) Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res 24: 1384–1395.
- Kajitani, R., D. Yoshimura, M. Okuno, Y. Minakuchi, H. Kagoshima, A. Fujiyama, K. Kubokawa, Y. Kohara, A. Toyoda and T. Itoh (2019) Platanus-allee is a de novo haplotype assembler enabling a comprehensive access to divergent heterozygous regions. Nat Commun 10: 1–15.
- Kim, D., J.M. Paggi, C. Park, C. Bennett and S.L. Salzberg (2019) Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37: 907–915.
- Kolmogorov, M., J. Yuan, Y. Lin and P.A. Pevzner (2019) Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol 37: 540–546.
- Kono, N. and K. Arakawa (2019) Nanopore sequencing: Review of potential applications in functional genomics. Dev Growth Differ 61: 316–326.
- Kovaka, S., A.V. Zimin, G.M. Pertea, R. Razaghi, S.L. Salzberg and M. Pertea (2019) Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20: 278.
- Krumsiek, J., R. Arnold and T. Rattei (2007) Gepard: A rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23: 1026–1028.
- Leisner, C.P., J.P. Hamilton, E. Crisovan, N.C. Manrique-Carpintero, A.P. Marand, L. Newton, G.M. Pham, J. Jiang, D.S. Douches, S.H. Jansky et al. (2018) Genome sequence of M6, a diploid inbred clone of the high-glycoalkaloid-producing tuber-bearing potato species Solanum chacoense, reveals residual heterozygosity. Plant J 94: 562–570.
- Li, G., J. Shao, Y. Wang, T. Liu, Y. Tong, S. Jansky, C. Xie, B. Song and X. Cai (2022) Rychc confers extreme resistance to Potato virus Y in potato. Cells 11: 2577.
- Li, H., B. Handsaker, A. Wysoker, T. Fennell, J. Ruan, N. Homer, G. Marth, G. Abecasis and R. Durbin (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079.
- Marijon, P., R. Chikhi and J.S. Varré (2020) Yacrd and fpa: Upstream tools for long-read genome assembly. Bioinformatics 36: 3894–3896.
- Mori, K., Y. Sakamoto, N. Mukojima, S. Tamiya, T. Nakao, T. Ishii and K. Hosaka (2011) Development of a multiplex PCR method for simultaneous detection of diagnostic DNA markers of five disease and pest resistance genes in potato. Euphytica 180: 347–355.
- Mori, K., N. Mukojima, T. Nakao, S. Tamiya, Y. Sakamoto, N. Sohbaru, K. Hayashi, H. Watanuki, K. Nara, K. Yamazaki et al. (2012) Germplasm release: Saikai 35, a male and female fertile breeding line carrying Solanum phureja-derived cytoplasm and potato cyst nematode resistance (H1) and Potato Virus Y resistance (Rychc) genes. Am J Potato Res 89: 63–72.
- Nie, X., F. Lalany, V. Dickison, D. Wilson, M. Singh, D. de Koeyer and A. Murphy (2016) Detection of molecular markers linked to Ry genes in potato germplasm for marker-assisted selection for extreme resistance to PVY in AAFC’s potato breeding program. Can J Plant Sci 96: 737–742.
- Ogawa, T., Y. Tomitaka, A. Nakagawa and K. Ohshima (2008) Genetic structure of a population of Potato virus Y inducing potato tuber necrotic ringspot disease in Japan; comparison with North American and European populations. Virus Res 131: 199–212.
- Ogawa, T., A. Nakagawa, T. Hataya and K. Ohshima (2012) The genetic structure of populations of Potato virus Y in Japan; Based on the analysis of 20 full genomic sequences. J Phytopathol 160: 661–673.
- Ohki, T., M. Sano, K. Asano, T. Nakayama and T. Maoka (2018) Effect of temperature on resistance to Potato virus Y in potato cultivars carrying the resistance gene Rychc. Plant Pathol 67: 1629–1635.
- Paluchowska, P., J. Śliwka and Z. Yin (2022) Late blight resistance genes in potato breeding. Planta 255: 127.
- Peloquin, S.J., A.C. Gabert and O. Rodomiro (1996) Nature of “Pollinator” effect in potato (Solanum tuberosum L.) haploid production. Ann Bot 77: 539–542.
- Pertea, G. and M. Pertea (2020) GFF Utilities: GffRead and GffCompare. F1000Res 9: 304.
- Pham, G.M., J.P. Hamilton, J.C. Wood, J.T. Burke, H. Zhao, B. Vaillancourt, S. Ou, J. Jiang and C.R. Buell (2020) Construction of a chromosome-scale long-read reference genome assembly for potato. Gigascience 9: giaa100.
- Ritter, E., T. Debener, A. Barone, F. Salamini and C. Gebhardt (1991) RFLP mapping on potato chromosomes of two genes controlling extreme resistance to potato virus X (PVX). Mol Gen Genet 227: 81–85.
- Rodriguez-Rodriguez, M., M. Chikh-Ali, S.B. Johnson, S.M. Gray, N. Malseed, N. Crump and A.V. Karasev (2020) The recombinant Potato virus Y (PVY) strain, PVY NTN, identified in potato fields in Victoria, Southeastern Australia. Plant Dis 104: 3110–3114.
- Sato, M., K. Nishikawa, K. Komura and K. Hosaka (2006) Potato Virus Y resistance gene, Rychc, mapped to the distal end of potato chromosome 9. Euphytica 149: 367–372.
- Schatz, M.C., J. Witkowski and W.R. McCombie (2012) Current challenges in de novo plant genome sequencing and assembly. Genome Biol 13: 243.
- Shirasawa, K., H. Hirakawa and S. Isobe (2016) Analytical workflow of double-digest restriction site-associated DNA sequencing based on empirical and in silico optimization in tomato. DNA Res 23: 145–153.
- Simão, F.A., R.M. Waterhouse, P. Ioannidis, E.V. Kriventseva and E.M. Zdobnov (2015) BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212.
- Sun, H., W.B. Jiao, K. Krause, J.A. Campoy, M. Goel, K. Folz-Donahue, C. Kukat, B. Huettel and K. Schneeberger (2022) Chromosome-scale and haplotype-resolved genome assembly of a tetraploid potato cultivar. Nat Genet 54: 342–348.
- Szajko, K., D. Strzelczyk-Zyta and W. Marczewski (2014) Ny-1 and Ny-2 genes conferring hypersensitive response to potato virus Y (PVY) in cultivated potatoes: Mapping and marker-assisted selection validation for PVY resistance in potato breeding. Mol Breed 34: 267–271.
- Takagi, H., A. Abe, K. Yoshida, S. Kosugi, S. Natsume, C. Mitsuoka, A. Uemura, H. Utsushi, M. Tamiru, S. Takuno et al. (2013) QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 74: 174–183.
- Takeuchi, T., J. Sasaki, T. Suzuki, H. Horita and S. Iketani (2008) High-resolution maps and DNA markers of the Potato virus Y resistance gene Rychc and the potato cyst nematode resistance gene H1. Breed Res 10 (Suppl. 1): 148 (in Japanese).
- Tang, D., Y. Jia, J. Zhang, H. Li, L. Cheng, P. Wang, Z. Bao, Z. Liu, S. Feng, X. Zhu et al. (2022) Genome evolution and diversity of wild and cultivated potatoes. Nature 606: 535–541.
- Van Breukelen, E.W.M., M.S. Ramanna and J.G.T. Hermsen (1977) Parthenogenetic monohaploids (2n = x = 12) from Solanum tuberosum L. and S. verrucosum Schlechtd. and the production of homozygous potato diploids. Euphytica 26: 263–271.
- Van der Vossen, E.A.G., J.N.A.M.R. van der Voort, K. Kanyuka, A. Bendahmane, H. Sandbrink, D.C. Baulcombe, J. Bakker, W.J. Stiekema and R.M. Klein-Lankhorst (2000) Homologues of a single resistance-gene cluster in potato confer resistance to distinct pathogens: A virus and a nematode. Plant J 23: 567–576.
- Van Lieshout, N., A. van der Burgt, M.E. de Vries, M. ter Maat, D. Eickholt, D. Esselink, M.P.W. van Kaauwen, L.P. Kodde, R.G.F. Visser, P. Lindhout et al. (2020) Solyntus, the new highly contiguous reference genome for potato (Solanum tuberosum). G3 (Bethesda) 10: 3489–3495.
- Vaser, R., I. Sović, N. Nagarajan and M. Šikić (2017) Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27: 737–746.
- Walker, B.J., T. Abeel, T. Shea, M. Priest, A. Abouelliel, S. Sakthikumar, C.A. Cuomo, Q. Zeng, J. Wortman, S.K. Young et al. (2014) Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9: e112963.
- Xu, X., S. Pan, S. Cheng, B. Zhang, D. Mu, P. Ni, G. Zhang, S. Yang, R. Li, J. Wang et al. (2011) Genome sequence and analysis of the tuber crop potato. Nature 475: 189–195.
- Yamakawa, H., E. Haque, M. Tanaka, H. Takagi, K. Asano, E. Shimosaka, K. Akai, S. Okamoto, K. Katayama and S. Tamiya (2021) Polyploid QTL‐seq towards rapid development of tightly linked DNA markers for potato and sweetpotato breeding through whole-genome resequencing. Plant Biotechnol J 19: 2040–2051.
- Zhang, Z., S. Schwartz, L. Wagner and W. Miller (2000) A greedy algorithm for aligning DNA sequences. J Comput Biol 7: 203–214.
- Zhou, Q., D. Tang, W. Huang, Z. Yang, Y. Zhang, J.P. Hamilton, R.G.F. Visser, C.W.B. Bachem, C.R. Buell, Z. Zhang et al. (2020) Haplotype-resolved genome analyses of a heterozygous diploid potato. Nat Genet 52: 1018–1023.