Review
Biomolecular Structure Model Evaluation and Modeling for cryo-EM maps

2023 Volume 4 Issue 2 Pages 91-103
Details
2023 Volume 4 Issue 2 Pages 91-103
2015年以降、急速に発展してきたクライオ電子顕微鏡(cryo-Electron Microscopy, cryo-EM)は、タンパク質やDNA、RNAといった生体分子の三次元構造の解析において、欠かすことのできない技術となっている。一方、最近では人工知能や深層学習、画像認識技術などの計算科学の手法が多くの科学分野で適用され、著しい成果を挙げている。最近ではこれら2つの技術の発展と融合により、生体分子の研究の基礎である構造生物学は、急速に大きく進展している。本総説では、まずクライオEMの背景と基本的な技術を紹介し、さらに利用可能なデータベースやデータ構造についても紹介する。その後、クライオEMデータにおける深層学習の適用について、特に生体分子構造の解析に焦点を当て、筆者らの研究グループが開発したプログラムを中心に紹介する。
クライオEM(cryo-Electron Microscopy)の「クライオ」は、「低温」を意味しており、測定するサンプルを低温で凍結させて透過電子顕微鏡(Transmission Electron Microscopy)でタンパク質や核酸などといった生体分子の構造データを解析する、というのが基本的な手法である[1, 2]。ではクライオEMから得られる構造データとはどのようなものだろうか?それを説明するためにクライオEMの原理を簡単に図1から紹介する。

クライオEMでは、まず測定するサンプルを厚さ20nm程度の薄い平面上で細かく分けられた各グリッドに入れ、水中に存在するような環境を保ったまま急速に凍結させる。次に、サンプルが入ったこの薄いフィルム状のグリッドを透過するように電子ビームが照射され、電子検出器(Electron Detector)が透過した二次元画像データを取得する。この二次元画像データには、各グリッド内に分布している生体分子の二次元投影画像が数千から数十万含まれている。この生体分子の二次元投影画像は、パーティクルと呼ばれており、個々のパーティクルはその生体分子が様々な方向を向いた二次元画像である。こうして得られたパーティクルを三次元空間上で合成して、生体分子の三次元構造が構築される[3]。この計算のことを3D reconstructionという。この計算は一つの三次元構造を様々な角度から写真を取り、その二次元の写真を元に三次元の形状を計算し、立体画像を得るのと基本的に同じである。二次元パーティクルから三次元構造を構築する計算には、cryoSPARC[4]、RELION[5]などのソフトウェアが主流で使われている。クライオEMの中でも近年発展が著しい分野の一つであり、深層学習やデータ生成技術を使った手法が多く発表されている[6, 7, 8]が、本総説では取り上げる内容から外れるのでMiolaneらの総説[6]を参照されたい。
このように、クライオEMから得られる生体分子の構造情報とは、生体分子の二次元投影画像(パーティクル)と、三次元構造データである。クライオEMにより取得した二次元投影画像のデータの一部は、EMPIAR (Electron Microscopy Public Image ARchive)[9]に登録される。後述するEMDBのエントリー数が2023年6月現在、27,501であるのに対し、EMPIARは1,320であり、利用できるデータは非常に限られている。ただし、EMPIARに登録される1エントリーのデータサイズは数百GBから数TBである。一方、三次元構造データはEMDB (Electron Microscopy Data Bank)[10]に4桁または5桁の数字で表記されるIDで登録される。三次元構造データは、基本的には三次元グリッドの各グリッド点に電子密度の数値が記録されたボリュームデータである。このボリュームデータは、クライオEMマップ、EMマップ、電顕マップ、EM密度マップ(cryo-EM map or EM density map)などと呼ばれる。生体分子の構造解析には主にクライオEMマップが用いられる。一部の解像度の高いクライオEMマップからは原子構造モデルが構築され、PDBデータベース(Protein Data Bank[11])に登録されている。2023年6月現在、PDBに登録されているエントリーのうち16,383の原子構造モデルはクライオEMマップから構築されている。
クライオEMには他の構造解析手法と比較して様々な利点がある。例えば、ウィルスの構造やリボソームといった測定する生体分子のサイズが大きい場合や、X線結晶解析では難しかった膜タンパク質の構造でも、結晶化する必要がないので構造解析が可能である。さらに、生体分子の動的構造変化を捉えることができる[8, 12]、などである。
2017年のノーベル化学賞はクライオEMの開発の功績についてであったが、クライオEMの主要な技術自体は1970〜80年代に開発されたものであった[13]。2010年代前半までは、X線結晶解析やNMRと比べて解像度において劣っていたため構造生物学の主流の手法にはなっていなかった。しかし2015年以降、電子検出器の解像度の向上、サンプルの精製技術の向上、そして大量の二次元パーティクルを処理する計算機の計算速度の向上と三次元構造を構築する計算アルゴリズムの改良といった数々の技術的進歩が組み合わさった結果、クライオEMマップの解像度はほぼ原子レベルの解像度まで到達可能になった[14]。図2は、各年に登録されたクライオEMマップの数と解像度の内訳を示している。2010年代前半までとそれ以降を比較するならば、登録されたクライオEMマップの数が2015年以降飛躍的に増えているのが分かる。これは前述した技術的進歩により、クライオEMマップの取得がより早く簡単になったことを反映している。また解像度に応じたエントリ数に着目するならば、4Åよりも高い解像度を持つクライオEMマップが現在主流となっていることがわかる。

クライオEMマップがどれだけ詳細な情報を持っているかを表す指標として「解像度」(resolution)が最もよく使われる。単位は10−10mのオングストローム(Å)である。解像度が高い(値(Å)が小さい)ほど、より詳細な形状がクライオEMマップから見られることを意味している。クライオEMマップの解像度を計算するには、まず同じ試料から得られたパーティクルのデータを半分ずつに分け、それぞれの独立した二次元投影画像データセットから個々にクライオEMマップを構築する。得られた2つのクライオEMマップはフーリエ変換され、その2つのデータ間のフーリエシェル相関係数(FSC, Fourier Shell Correlation)から解像度を推定される[15, 16]。2つの独立データから構築されたクライオEMマップが、どこまで詳細な部分まで一致しているかを周波数と相関係数のグラフから推定する。
さて、図3と図4でクライオEMマップの解像度とボリュームデータの形を比較してみた。1.75〜3.9Åの異なる解像度で解かれたapoferritin分子のクライオEMマップを4つ示した[17, 18]。図3はそれぞれのクライオEMマップの全体像を示している。一般的に、解像度が2Åあるとタンパク質の主鎖構造とアミノ酸残基の種類が判別できるほど側鎖構造が判別できる。解像度が3Åでは主鎖構造を観察することができるが、アミノ酸残基の種類を特定するのがやや難しくなってくる。解像度が4〜5Åの場合、タンパク質のαヘリックスとβシート構造ははっきりと観察できるものの、アミノ酸残基の側鎖は突起状の形状としか観察されない。

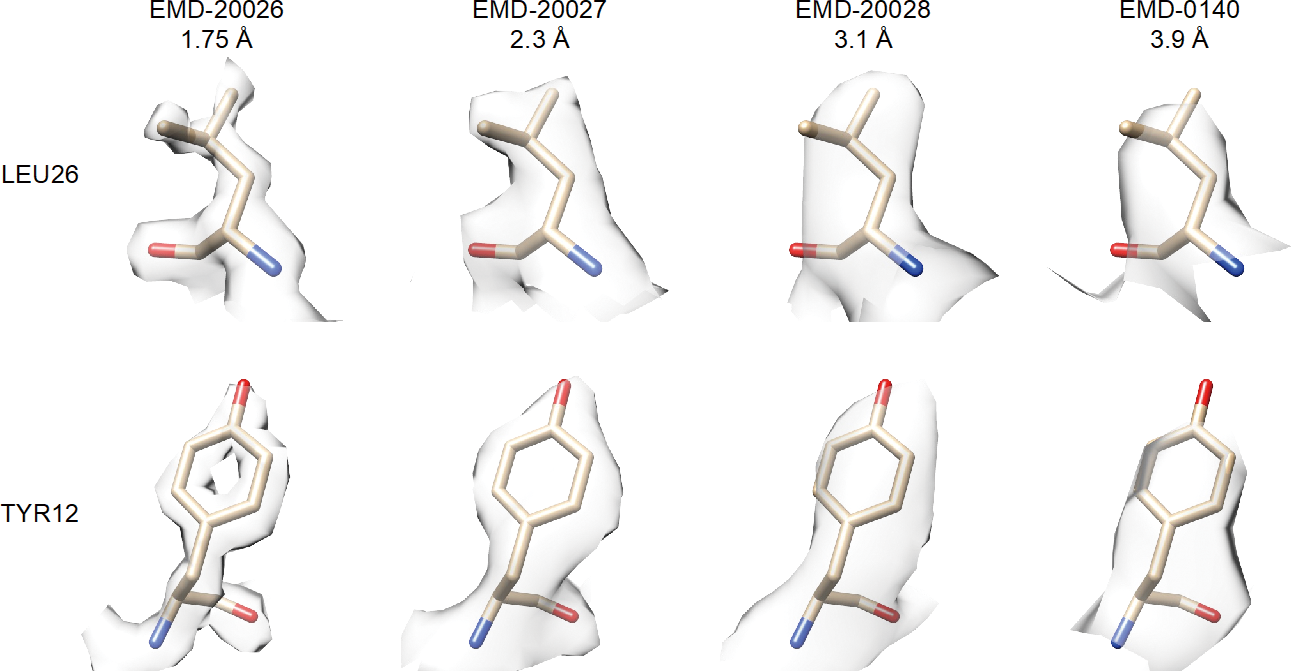
図4は、PDBに登録されているApoferritinの原子構造モデルPDB 3AJOと4つクライオEMマップの比較である。特徴的な形をもつ残基番号26のロイシン(Leucine, LEU26)と残基番号12のチロシン(Tyrosine, TYR12)の周辺を拡大表示している。4つのうち最も解像度が高いクライオEMマップ(EMD-20026) からは、タンパク質を構成する原子の形状がはっきりと確認できる。側鎖の枝分かれがある部分や、リングの穴も見ることができる。逆に4つのうち解像度が最も低いEMD-0140では、側鎖構造が突起状の形でのみ観察することができる。4Å程度の解像度であると、クライオEMマップの情報のみからチロシンとロイシンを判別するのは難しいことが分かるだろう。
解像度はEMマップの詳細度を示す指標として広く用いられているが、一つのクライオEMマップ全体の詳細度を一つの数値だけで表現するのは無理があることを留意しなくてはいけない。程度の違いはあれども、タンパク質は常に生体内で構造を変化させている。ドメイン構造の相対位置が大きく動くような構造変化から、一部のループ構造が動いているものまで様々なタンパク質の構造変化がタンパク質には起こっている。そのようなタンパク質構造中のフレキシブルな部分に相当する空間では、クライオEMマップからはっきりとした構造データが得られないことが多い。したがって、解像度が2Åと報告されているクライオEMマップであったとしても、ある局所部分の解像度は低く不鮮明ということがあり得る。クライオEMマップの局所的な解像度を推定する手法として、フーリエシェル相関係数を使うものや深層学習を使うものが提案されており、詳しくはSorzanoの総説[21]を参照されたい。
ここまでで、クライオEMから得られるクライオEMマップにはどのような情報が含まれているのかを見ることができた。さらに解像度に応じて、そのクライオEMマップがどこまで詳細な情報を有しているかを知るための重要な指標であることを確認した。次のステップとして、クライオEMマップのデータから生体分子構造(タンパク質と核酸)の原子モデルを構築する手法、さらに原子モデルの精度を評価する手法に焦点を当ててみよう。
2.1 クライオEMマップに基づく生体分子の構造情報同定のための3次元畳み込みニューラルネットワーク(3DCNN)の適応クライオEMマップから生体分子構造のモデリングをするには、まずクライオEMマップのデータから生体分子の特徴を抽出する必要がある。クライオEMマップの解像度に応じて観察することができる生体分子の構造情報は大きく異なっていることから、生体分子構造情報の抽出という課題では様々なレベルでの問題設定が考えられる。例えば、①解像度5〜10ÅのクライオEMマップから、タンパク質の二次構造と核酸の場所を推定する。②解像度3〜5ÅのクライオEMマップから、アミノ酸の種類を推定する。③解像度2〜4ÅのクライオEMマップから、重原子の種類と、主鎖構造の繋がりを推定する。といったものである。図4で示したのは一例ではあるが、チロシンとロイシンという2つの異なるアミノ酸残基を区別するには、2〜3Å以上の解像度が必要となることがわかる。したがって、現在主流となっている3〜5Åの解像度を持つクライオEMマップから、注目している場所のアミノ酸残基を特定し、さらに全ての主鎖構造を正確に見つけるのは簡単ではないことが分かるだろう。
さて、クライオEMマップのデータ構造は、格子点1つに1つの値が記録されたグリッドデータである。クライオEMマップとは、いわば三次元のピクセルを持つ白黒画像のデータ構造と同じである。よって、クライオEMマップからタンパク質や核酸などの構造情報を同定するということは、白黒画像の中にどのような物体がどこに何が描かれているかを同定する画像認識問題と本質的に同じである。
画像認識問題は、計算科学の分野において深層学習の応用により飛躍的な進展がみられる分野である。特に、深層学習の代表的な手法である畳み込みニューラルネットワーク(CNN[22])が画像認識に広く用いられている。CNNは、画像の局所情報を複数のニューロン層を介して畳み込むことで、局所と広域の情報を効果的に学習する手法である。より多くの画像データと深いニューロン層を用いたCNNの訓練により、より高精度な画像の分類が可能となっている。
近年飛躍的に増えたEMマップデータは、深層学習のトレーニングに使えるデータが年々増えていることを意味している。そこで筆者らの研究グループは、3次元の畳み込みニューラルネットワーク(3DCNN[23])を使い、解像度5〜10ÅのクライオEMマップから、タンパク質の二次構造と核酸の場所を推定するアプローチを提案した[24, 25]。提案手法では、EMDBに登録されている中低解像度(5〜10Å)のクライオEMマップとPDBに登録されている原子モデルから約44万の格子点データを学習データセットとして抽出した。1つの格子点データは113の三次元配列であり、その格子点データの中央の座標に対応する生体分子の構造情報(二次構造情報など)がラベルとして紐付けられている。3DCNNは、格子点データから、対応する構造情報を予測するようにトレーニングされる。筆者らの研究グループは、トレーニングした3DCNNを活用して、クライオEMマップからタンパク質の二次構造を同定するプログラムEmap2sec[24]と、タンパク質の二次構造に加えてDNAとRNAの位置を同定するEmap2sec+[25]を公開した。2つのプログラムは、サーバー上で計算する形式(https://em.kiharalab.org)と、GitHubでのソースコードまたはオンライン計算プラットフォームのGoogle Colabで使用できる形式で公開されている(https://kiharalab.org/emsuites/)。
図5は、実際にクライオEMマップ(EMD-7103[26] 解像度5.5Å)を入力データとし、Emap2sec+サーバー(https://em.kiharalab.org/algorithm/emap2sec+)がタンパク質二次構造とDNA/RNAの場所を計算した例である。比較のため、PDBに登録されている原子構造モデル(PDB 6BJS)も示した。この例でEmap2sec+は73%の精度で二次構造とDNA/RNAを同定した。このように比較的低い解像度5〜10ÅのクライオEMマップからであっても、深層学習を使いタンパク質二次構造や核酸構造を同定することが可能である。

PDBに登録されている構造モデルのうち、クライオEMマップから構築された構造モデルのエントリー数は2023年現在1万6千以上である。PDBの総エントリー数約20万と比較すると数は少ないが、EMマップ数の増加と共にPDBに登録される構造モデルの数も増加傾向である。
さて、筆者らの研究グループはPDBに登録されているクライオEMマップから構築されたタンパク質構造モデルを解析し、同じアミノ酸配列を持っているにも関わらず対応するアミノ酸残基の位置が異なってしまっている構造モデルの存在を報告した[27]。図6は例としてPDB 6L54 C鎖とPDB 6Z3R C鎖の構造モデルを示している。この2つの構造モデルは、どちらもSMG9タンパク質であり、同じアミノ酸配列を持っている。それぞれは異なるクライオEMマップ(EMD-0837[28]とEMD-11063[29])から構築されたものである。図6が示すように2つの構造モデルの全体構造は非常によく似ているが、拡大表示した右側2つのパネルを見ると、同じ残基番号の同じアミノ酸であるフェニルアラニン421番(Phe421)、アスパラギン酸423番(Asp423)、そしてグルタミン酸425番(Glu425)の位置がPDB 6L54(シアン)と6Z3R(ピンク)で異なっていることが分かる。ワイヤーフレームで表示されたEMマップを見ると、どちらもアミノ酸側鎖の形状が観察でき、構造モデルとクライオEMマップは一致しているように見える。この2つの構造モデルの矛盾は、PDBに登録された構造モデルにエラーが存在していることを示唆している。このような構造モデリングのエラーを検出するには、熟練した構造生物学者の知見とスキルが求められる。そのため構造モデルの精度評価は、構造モデリングにおいて大きな課題の一つである。

クライオEMマップと構造モデルを比較し、構造モデルを評価する手法はこれまでいくつか提案されてきた[30, 31, 32, 33, 34]。それらの手法は、基本的にクライオEMマップの密度データと構造モデルが一致しているか、構造モデルはタンパク質らしい構造を取っているか、を評価する手法である。その中で代表的な手法の一つであるQ-score[18]は、最近PDBウェブサイトで原子モデルとクライオEMマップの一致度を評価するツールとして使用されるようになった。Q-scoreは、構造モデルの各原子の位置に注目し、Gaussian-like関数からその原子の周辺の密度データを推定する。この推定された密度データの分布と、実際にクライオEMマップから観察されるその原子周辺の密度データの分布を比較し類似度を計算する。2つ密度データの分布が異なると、Q-scoreは低い値を示す。これは、その原子周辺の解像度が低いか、原子モデルがEMマップとよく一致していない、ということを意味する。Q-scoreの値を解釈する際は、値はEMマップの解像度に大きく依存する点を留意する必要がある。Q-scoreの値が低いとしても、EMマップの解像度が低い場合と、構造モデルがクライオEMマップに一致していないという2つの原因が可能性としてある。
一方筆者らの研究グループは、クライオEMマップの密度データを直接扱うQ-scoreとは異なり、深層学習によって生体分子構造の特徴をクライオEMマップから計算し、その局所の特徴と構造モデルの一致度で構造モデルの精度を評価する手法DAQスコア[27]を開発した。筆者らの研究グループは、先に述べたEmap2sec+の手法を拡張し、クライオEMマップの格子点に相当する座標に存在する局所のタンパク質の特徴(原子の種類、20種類のアミノ酸、及び3種類の二次構造)の存在確率を予測するように3DCNNの学習を行った。学習用データは解像度が2.5Åから5.0ÅのEMマップを使用した。図7はDAQスコアの計算フローを示している。構造モデルの評価の際は、113Å3の格子点データがクライオEMマップからスキャンされ、格子点データから3DCNNが算出した確率と、対応する座標に存在する構造モデルの特徴を比較する。DAQスコアの計算には、ランダムに座標を選んだ場合に推定される確率の平均値と注目する場所の確率を使ったログオッズスコアが使用されている。こうして計算されたDAQスコアは構造モデルのCα原子について計算される。筆者らの研究グループの報告によると、DAQスコアは構造モデルの評価において、既存の手法と比較して最も高いエラー検出能力を示している。

図8でクライオEMマップ(EMD-22458) と構造モデル(PDB 7JSN[35])におけるDAQスコアの計算結果を示した。ちなみにDAQスコアの訓練用データにはこの構造モデルおよび類縁タンパク質は含まれていない。この構造モデルは最初2020年10月にPDBデータベースに登録され、2021年に同じ研究グループによって更新されている。この構造モデルや2つのバージョンの構造モデル(図8bとc)を見ると、大部分が同じ主鎖構造をしているが、図8dで示したようにアミノ酸残基の位置が変更されているのが分かる。この2つのバージョンの構造モデルに対しDAQスコアを計算したところ、ver1.1で多くの箇所が負のDAQスコアを示し(図8e)、その位置はver2.0で修正された場所と一致していた。よってこの例では、DAQスコアはver1.1の構造モデルのどの箇所でエラーがあるかを判別できており、研究者グループが構造をアップデートしたのと同じ場所を指摘している。DAQスコアのソースコードとGoogle Colab上で動作するプログラムはGitHubから公開されている: https://github.com/kiharalab/DAQ

深層学習を使ったDAQスコアにより、構造モデルのエラーを検出できるようになった。では当然考えられる疑問として、現在公開されているPDBデータベースの構造モデル中に、どれだけのエラーが含まれているのだろうか?筆者らの研究グループは、PDBデータベースに登録されている構造モデルのうち、解像度が2.5Åから5.0ÅのクライオEMマップからモデリングされた構造モデルに対してDAQスコアの計算を行い、データベースとして公開した[36]。バージョンの違いも考慮した167,288個のタンパク質鎖の構造モデルのDAQスコアがDAQスコアデータベース(https://daqdb.kiharalab.org)で公開されている(図9)。筆者らの研究グループの報告[36]では、約8%のタンパク質鎖の構造モデルにおいて、アミノ酸残基のアサイメントのエラーが少なくとも1つ以上存在していることがDAQスコアによって指摘されている。PDBデータベースに登録された構造モデルがクライオEMマップによって構築されている場合、DAQスコアで個々のモデルの精度を確認するならば、精度の低い構造モデルを使用することを避けられるだろう。

クライオEMマップからタンパク質構造をモデリングする手法は、密度データを直接取り扱う従来手法と深層学習を使うものに大別できる。前者のうちPathwalking[37]とMAINMAST[38]は密度データに沿ってタンパク質主鎖構造をトレースする手法であり phenix.map_to_model[39](モデリングツールPhenix内に含まれる)とRosetta[40, 41]は、密度データに一致するようにタンパク質のフラグメント構造モデルをEMマップにフィッティングさせてモデリングを行うというものである。後者の深層学習を使う手法の中で、Deeptracer[42]は画像のセグメンテーションに使われるU-Netsを使いタンパク質の主鎖構造とアミノ酸の種類を同定してモデリングをする。phenix.dock_and_rebuild[43]は、AlphaFold2[44]モデル中の信頼度が高い部分をEMマップに重ね合わせて、さらにEMマップとモデルの一致度合いを高めるように構造モデルの修正を繰り返す手法である。
筆者らの研究グループは、DAQスコアで開発した深層学習による局所の構造特徴を予測する手法をモデリングに応用し、さらにDAQスコアを構造最適化の目的関数に使用するDeepMainmast[45]を公開した。図10はDeepMainmastによるモデリングの流れを示している。DeepMainmastの計算は、主に以下の3つのステップから構成される。(1)EMマップから深層学習を使い、20種のアミノ酸と原子種の存在確率を計算する。(2)主鎖原子の存在確率から経路最適化手法を使い主鎖構造をトレーシングする。(3)アミノ酸配列を主鎖構造に当てはめ、得られたフラグメント構造をDAQスコアが最も高くなるように組み合わせる。

DeepMainmastの特徴の一つはAlphaFold2モデルを追加の構造フラグメントとして使用できることである。クライオEMマップのデータが不鮮明であったとしても、周辺の主鎖構造とAlphaFold2モデルを比較して、欠けている部分を埋め合わせることができる。これにより、更にモデリングの精度を向上させることが可能である。DeepMainmastのプログラムとGoogle Colabバージョンはhttps://github.com/kiharalab/DeepMainMastにて公開されている。
2.5 クライオEMマップからDNA/RNA構造のモデリングDNA/RNAといった核酸構造は、タンパク質構造と共にクライオEMで多く解析されている生体分子構造である。しかしながらクライオEMマップからの核酸構造のモデリングは、タンパク質構造のモデリングほど研究が行われていない。核酸構造モデルの多くは、Coot[46]やUCSF Chimera[19]といった3Dグラフィック画面上での手動のモデリングツールによって構築される。そのため深層学習によって今後大きく発展することが期待されている分野である。
前述したEmap2sec+は、3DCNNを使いEMマップから核酸の場所を見つけ出す手法である。筆者らの研究グループはこの手法を拡張し、DeepMainmastの構造モデリング手法を組み合わせて核酸構造のモデリングをするプログラムCryoREAD[47]を公開した。図11はCryoREADの計算フローを示している。CryoREADはまず深層学習によって核酸構造の主鎖を構成するリン酸基、糖(リボースまたはデオキシリボース)そして塩基の場所を予測する。深層学習は、格子点上における塩基の種類の確率を同時に計算する。次にCryoREADは、深層学習から得られた確率情報を使い、塩基データがアサインされたフラグメント構造を多数計算する。最後にフラグメント構造を組み合わせて核酸構造モデルを構築する。筆者らのベンチマークによると、CryoREADは現在使用できる従来手法のモデリングツールphenix.map_to_model[39]と比較して、明らかに高い主鎖構造のCoverageと塩基の種類の予測精度を達成した。CryoREADのプログラムとGoogle Colabバージョンがhttps://github.com/kiharalab/CryoREADで公開されている。

近年、増加傾向にあるクライオEMマップの解析に、深層学習を応用した構造モデル評価法や、タンパク質・核酸構造モデリングの新たな手法を紹介した。クライオEMマップは、深層学習の得意とする画像解析を直接適用できる分野であるため、今後ますます発展することが見込まれる。最近では、画像生成モデルによって作成したい画像データを自由に作り出すことができるようになっている。クライオEMマップは構造モデルと比較して多くは解像度が低く、ぼやけた画像であると言える。生成モデルにより、クライオEMマップがより詳細な構造データを示すように改変できるようになるだろう。しかし、そのような生成されたクライオEMマップは実験によって観察されたデータではないため詳細であっても誤った構造データになりうる事に注意が必要となる。例えば、アミノ酸配列から直接タンパク質構造をモデリングするAlphaFold2などの結果を生成モデルに教えることで、目的のアミノ酸配列と整合性のある詳細なクライオEMマップを生成することが可能となるだろう。
筆者らの研究グループでは深層学習をクライオEMデータに適用した手法を開発している。本稿で紹介した手法は、Webサイト(https://kiharalab.org/web/software)で公開しているものである。クライオ電子顕微鏡に関連したソフトウェアは、Cryo-EM Suite(https://kiharalab.org/emsuites/)内で個別に公開している。これらのソフトウェアの多くは、複雑な設定を必要とせず、インターネットブラウザー上で動作するGoogle Colab版も公開している。クライオEMに関連した研究を行っている研究者に、手軽にこれらのツールを活用していただきたいと考えている。
 |
寺師 玄記 2008年に北里大学薬学部で博士(薬学)を取得。2017年まで同大学で講師を務めた後、渡米しパデュー大学理学部生物科学科で博士研究員となる。2021年より、同大学でAssistant Research Scientistとして研究を行う。クライオEMとタンパク質構造モデリングに関連した研究に従事。 |
 |
木原 大亮 1999年に京都大学理学部で博士(理学)を取得。2003年までの米国での博士研究員を経て、2003年にAssistant Professorとしてパデュー大学に赴任。以後、同大学で2009年にAssociate Professor、2014年に教授(現職)。タンパク質の構造、機能に関するバイオインフォマティクスの研究に従事。 |